ПРАВИТЕЛЬСТВО РОССИЙСКОЙ ФЕДЕРАЦИИ
ПОСТАНОВЛЕНИЕ
от 6 июля 2012 г. N 686
ОБ УТВЕРЖДЕНИИ ПОЛОЖЕНИЯ
О ЛИЦЕНЗИРОВАНИИ ПРОИЗВОДСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
В соответствии с Федеральным законом "О лицензировании отдельных видов деятельности" Правительство Российской Федерации постановляет:
1. Утвердить прилагаемое Положение о лицензировании производства лекарственных средств.
2. Признать утратившим силу постановление Правительства Российской Федерации от 3 сентября 2010 г. N 684 "Об утверждении Положения о лицензировании производства лекарственных средств" (Собрание законодательства Российской Федерации, 2010, N 37, ст. 4698).
Председатель Правительства
Российской Федерации
Д.МЕДВЕДЕВ
Утверждено
постановлением Правительства
Российской Федерации
от 6 июля 2012 г. N 686
ПОЛОЖЕНИЕ
О ЛИЦЕНЗИРОВАНИИ ПРОИЗВОДСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
1. Настоящее Положение устанавливает порядок лицензирования деятельности по производству лекарственных средств, осуществляемой юридическими лицами в соответствии с Федеральным законом "Об обращении лекарственных средств".
2. Лицензирование деятельности по производству лекарственных средств для медицинского применения осуществляет Министерство промышленности и торговли Российской Федерации, для ветеринарного применения - Федеральная служба по ветеринарному и фитосанитарному надзору (далее - лицензирующие органы).
3. Деятельность по производству лекарственных средств включает в себя работы по перечню согласно приложению N 1.
4. Лицензионными требованиями, предъявляемыми к соискателю лицензии на осуществление деятельности по производству лекарственных средств (далее - лицензия), являются:
а) наличие у соискателя лицензии помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, принадлежащих ему на праве собственности или на ином законном основании, необходимых для выполнения заявляемых работ, соответствующих установленным требованиям;
б) соответствие производства лекарственных средств для ветеринарного применения правилам надлежащей производственной практики в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств";
б(1)) соответствие производства лекарственных средств для медицинского применения правилам надлежащей производственной практики Евразийского экономического союза;
в) наличие в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств" промышленных регламентов, утвержденных руководителем производителя лекарственных средств (соискателя лицензии) и включающих в себя перечень используемых фармацевтических субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании и описание технологического процесса и методов контроля на всех этапах производства лекарственных средств;
г) наличие в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств" уполномоченного лица производителя лекарственных средств для ветеринарного применения, которое при вводе лекарственных средств для ветеринарного применения в гражданский оборот осуществляет подтверждение соответствия лекарственных средств для ветеринарного применения требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства для ветеринарного применения произведены в соответствии с правилами надлежащей производственной практики, имеет образование и стаж работы, соответствующие требованиям, установленным статьей 45 Федерального закона "Об обращении лекарственных средств", при производстве лекарственных средств для ветеринарного применения и аттестовано в порядке, установленном Министерством сельского хозяйства Российской Федерации;
г(1)) наличие уполномоченного лица (уполномоченных лиц) производителя лекарственных средств для медицинского применения, являющегося его работником, которое при вводе лекарственных средств в гражданский оборот осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза, которое аттестовано и включено в реестр уполномоченных лиц производителей лекарственных средств Евразийского экономического союза в соответствии с правом Евразийского экономического союза;
д) наличие работников, заключивших трудовые договоры, имеющих соответственно высшее или среднее профессиональное фармацевтическое, химическое, химико-технологическое, биологическое, биотехнологическое, медицинское или ветеринарное образование, ответственных за производство, маркировку и контроль качества лекарственных средств.
4(1). При намерении соискателя лицензии осуществлять производство фармацевтической субстанции спирта этилового (этанола) наряду с лицензионными требованиями, указанными в пункте 4 настоящего Положения, предъявляются следующие лицензионные требования:
а) оснащение емкостей для приемки этилового спирта автоматическими средствами измерения и учета концентрации и объема безводного спирта в этиловом спирте, объема этилового спирта;
б) программно-аппаратные средства должны обеспечивать прием и передачу информации о концентрации и об объеме безводного спирта в этиловом спирте, используемом для производства фармацевтической субстанции спирта этилового (этанола), объеме этого этилового спирта, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в этиловом спирте, объема этилового спирта;
в) оснащение основного технологического оборудования автоматическими средствами измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола);
г) программно-аппаратные средства должны обеспечивать прием и передачу информации, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола), об объеме ее производства, поставки и (или) использования для собственных нужд;
д) наличие на праве собственности, хозяйственного ведения или оперативного управления основного технологического оборудования для производства этилового спирта, зарегистрированного в соответствии со статьей 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
е) емкости для приемки этилового спирта для производства фармацевтической субстанции спирта этилового (этанола) должны быть соединены коммуникациями, отвечающими требованиям, установленным федеральным органом исполнительной власти, уполномоченным по контролю и надзору в области производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции, с основным технологическим оборудованием для производства спирта этилового (этанола).
4(2). Лицензионными требованиями, предъявляемыми к соискателю лицензии при намерении осуществлять производство спиртосодержащих лекарственных препаратов, а также производство других лекарственных средств с использованием фармацевтической субстанции спирта этилового (этанола) наряду с лицензионными требованиями, указанными в пункте 4 настоящего Положения, являются:
а) оснащение емкостей для приемки фармацевтической субстанции спирта этилового (этанола) автоматическими средствами измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола);
б) программно-аппаратные средства для производства спиртосодержащих лекарственных препаратов, а также в процессе производства других лекарственных средств должны обеспечивать прием и передачу информации, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола), об объеме ее закупки;
в) оснащение оборудования для учета объема оборота и использования фармацевтической субстанции спирта этилового (этанола) для производства спиртосодержащих лекарственных препаратов, а также в процессе производства других лекарственных средств с использованием фармацевтической субстанции спирта этилового (этанола) техническими средствами фиксации и передачи информации об объеме производства и оборота спиртосодержащей продукции в единую государственную автоматизированную информационную систему учета объема производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции (далее - единая государственная автоматизированная информационная система);
г) программно-аппаратные средства для производства спиртосодержащих лекарственных препаратов, а также в процессе производства других лекарственных средств должны обеспечивать прием и передачу информации об объеме использования фармацевтической субстанции спирта этилового (этанола), а также информации об объеме производства и (или) оборота (за исключением розничной продажи) спиртосодержащих лекарственных препаратов, полученной с применением технических средств фиксации и передачи информации об объеме производства и оборота спиртосодержащей продукции, в единую государственную автоматизированную информационную систему.
4(3). Требования подпунктов "в" и "г" пункта 4(2) настоящего Положения не распространяются на соискателей лицензии при намерении осуществлять производство спиртосодержащих лекарственных препаратов, включенных в перечень спиртосодержащих лекарственных препаратов, утвержденный в соответствии с абзацем третьим пункта 4 статьи 1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции".
5. Лицензионными требованиями, предъявляемыми к лицензиату при осуществлении деятельности по производству лекарственных средств, являются:
а) наличие у лицензиата помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, принадлежащих ему на праве собственности или на ином законном основании, необходимых для выполнения заявленных работ, соответствующих установленным требованиям;
б) соблюдение в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств" требований промышленных регламентов, утвержденных руководителем производителя лекарственных средств (лицензиата) и включающих в себя перечень используемых фармацевтических субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании и описание технологического процесса и методов контроля на всех этапах производства лекарственных средств;
в) в отношении производства лекарственных средств для ветеринарного применения соблюдение в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств" правил надлежащей производственной практики;
в(1)) в отношении производства лекарственных средств для медицинского применения соблюдение Правил надлежащей производственной практики Евразийского экономического союза;
г) наличие в соответствии со статьей 45 Федерального закона "Об обращении лекарственных средств" уполномоченного лица производителя лекарственных средств для ветеринарного применения, которое при вводе лекарственных средств для ветеринарного применения в гражданский оборот осуществляет подтверждение соответствия лекарственных средств для ветеринарного применения требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства для ветеринарного применения произведены в соответствии с правилами надлежащей производственной практики, а также которое имеет образование и стаж работы, соответствующие требованиям, установленным статьей 45 Федерального закона "Об обращении лекарственных средств", при производстве лекарственных средств для ветеринарного применения и аттестовано в порядке, установленном Министерством сельского хозяйства Российской Федерации;
г(1)) наличие уполномоченного лица (уполномоченных лиц) производителя лекарственных средств для медицинского применения, являющееся его работником, которое при вводе лекарственных средств в гражданский оборот осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями Правил надлежащей производственной практики Евразийского экономического союза, которое аттестовано и включено в реестр уполномоченных лиц производителей лекарственных средств Евразийского экономического союза в порядке, установленном Евразийской экономической комиссией. При назначении нового уполномоченного лица производителя лекарственных средств лицензиат обязан в течение 10 рабочих дней уведомить лицензирующий орган о фамилии, имени и отчестве (при наличии) уполномоченного лица и дате его назначения;
д) наличие работников, заключивших трудовые договоры, имеющих высшее или среднее профессиональное фармацевтическое, химическое, химико-технологическое, биологическое, биотехнологическое, медицинское или ветеринарное образование, ответственных за производство, маркировку и контроль качества лекарственных средств;
е) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" о запрете производства лекарственных средств, не включенных в государственный реестр лекарственных средств, за исключением лекарственных средств, производимых для проведения клинических исследований и экспорта, а также о запрещении производства фальсифицированных лекарственных средств, лекарственных средств для ветеринарного применения с нарушением правил надлежащей производственной практики, лекарственных средств для медицинского применения с нарушением Правил надлежащей производственной практики Евразийского экономического союза;
ж) соблюдение лицензиатом требований статьи 57 Федерального закона "Об обращении лекарственных средств" о запрете продажи недоброкачественных лекарственных средств, фальсифицированных лекарственных средств и контрафактных лекарственных средств;
з) соблюдение правил хранения лекарственных средств в соответствии со статьей 58 Федерального закона "Об обращении лекарственных средств";
и) соблюдение Правил уничтожения недоброкачественных лекарственных средств, фальсифицированных лекарственных средств и контрафактных лекарственных средств, утвержденных в соответствии со статьями 47 и 59 Федерального закона "Об обращении лекарственных средств", за исключением лекарственных средств для медицинского применения;
к) утратил силу. - Постановление Правительства РФ от 22.12.2020 N 2206;
к(1)) соблюдение правил регистрации операций, связанных с обращением лекарственных средств для медицинского применения, включенных в перечень лекарственных средств для медицинского применения, подлежащих предметно-количественному учету, в специальных журналах учета операций, связанных с обращением лекарственных средств для медицинского применения, и правил ведения и хранения специальных журналов учета операций, связанных с обращением лекарственных средств для медицинского применения;
л) повышение квалификации лиц, указанных в подпунктах "г", "г(1)" и "д" настоящего пункта, не реже 1 раза в 5 лет;
м) регистрация в системе мониторинга движения лекарственных препаратов для медицинского применения;
н) соблюдение требований статьи 67 Федерального закона "Об обращении лекарственных средств":
об обеспечении в порядке и в составе, которые установлены Правительством Российской Федерации, внесения информации о лекарственных препаратах для медицинского применения в систему мониторинга движения лекарственных препаратов для медицинского применения;
о нанесении для целей идентификации упаковок лекарственных препаратов для медицинского применения в порядке, установленном Правительством Российской Федерации, на первичную упаковку (в отношении лекарственных препаратов для медицинского применения, для которых не предусмотрена вторичная упаковка) и вторичную (потребительскую) упаковку лекарственных препаратов для медицинского применения средства идентификации, за исключением лекарственных препаратов для медицинского применения, производимых для проведения клинических исследований, экспорта, лекарственных препаратов, указанных в частях 5 и 8 статьи 13 указанного Федерального закона, радиофармацевтических лекарственных препаратов, пиявок медицинских и газов медицинских.
5(1). Лицензионными требованиями, предъявляемыми к лицензиату при осуществлении производства фармацевтической субстанции спирта этилового (этанола), наряду с лицензионными требованиями, указанными в пункте 5 настоящего Положения, являются:
а) оснащение емкостей для приемки этилового спирта у организаций, использующих этиловый спирт для производства фармацевтической субстанции спирта этилового (этанола), автоматическими средствами измерения и учета концентрации и объема безводного спирта в этиловом спирте, объема этилового спирта;
б) программно-аппаратные средства организаций, использующих этиловый спирт для производства фармацевтической субстанции спирта этилового (этанол), должны обеспечивать прием и передачу информации о концентрации и об объеме безводного спирта в этиловом спирте, объеме этилового спирта, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в этиловом спирте, объема этилового спирта;
в) оснащение основного технологического оборудования организаций, осуществляющих производство фармацевтической субстанции спирта этилового (этанола), автоматическими средствами измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола);
г) программно-аппаратные средства организаций, осуществляющих производство фармацевтической субстанции спирта этилового (этанола), должны обеспечивать прием и передачу информации, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола), об объеме ее производства, поставки и (или) использования для собственных нужд;
д) наличие на праве собственности, хозяйственного ведения или оперативного управления основного технологического оборудования для производства этилового спирта, зарегистрированного в соответствии со статьей 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
е) емкости для приемки этилового спирта для производства фармацевтической субстанции спирта этилового (этанола) должны быть соединены с коммуникациями, отвечающими требованиям, установленным федеральным органом исполнительной власти, уполномоченным по контролю и надзору в области производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции, с основным технологическим оборудованием для производства спирта этилового (этанола);
ж) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" по производству фармацевтической субстанции спирта этилового (этанола) методом разведения водой очищенной ректификованного этилового спирта из пищевого сырья, произведенного на основании лицензии на производство этилового спирта для производства фармацевтической субстанции спирта этилового (этанола) в соответствии с требованиями Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции", и по месту осуществления производства фармацевтической субстанции спирта этилового (этанола);
з) соблюдение лицензиатом требований о запрете производства спиртосодержащих лекарственных препаратов по месту осуществления производства фармацевтической субстанции спирта этилового (этанола);
и) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" о запрете производства фармацевтической субстанции спирта этилового (этанола) на основном технологическом оборудовании для производства этилового спирта, указанном в пункте 1.1 статьи 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
к) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" о запрете реализации (передачи в установленном законодательством Российской Федерации порядке) фармацевтической субстанции спирта этилового (этанола) организациям оптовой торговли лекарственными средствами;
л) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" об осуществлении перевозки фармацевтической субстанции спирта этилового (этанола) с соблюдением требований, установленных статьей 9 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
м) соблюдение лицензиатом требований о реализации (передаче в установленном законодательством Российской Федерации порядке) фармацевтической субстанции спирта этилового (этанола) производителям лекарственных средств для производства лекарственных средств в емкостях объемом не более 1 литра и (или) не менее 1000 литров, а организациям, указанным в пунктах 3 - 5 части 8 статьи 45 Федерального закона "Об обращении лекарственных средств", - в таре объемом не более 1 литра;
н) соблюдение лицензиатом требований о запрете реализации (передачи в установленном законодательством Российской Федерации порядке) фармацевтической субстанции спирта этилового (этанола) организациям и индивидуальным предпринимателям, осуществляющим разведение, выращивание и содержание животных;
о) соблюдение лицензиатом требований о запрете производства фармацевтической субстанции спирта этилового (этанола) при прекращении или аннулировании действия лицензии на производство этилового спирта для производства фармацевтической субстанции спирта этилового (этанола).
5(2). Лицензионными требованиями, предъявляемыми к лицензиату при осуществлении производства спиртосодержащих лекарственных препаратов исходя из объема потребительской тары (упаковки) и (или) стоимости и (или) функционального назначения спиртосодержащих лекарственных препаратов, а также производства других лекарственных средств с использованием фармацевтической субстанции спирта этилового (этанола) наряду с лицензионными требованиями, указанными в пункте 5 настоящего Положения, являются:
а) оснащение емкостей для приемки фармацевтической субстанции спирта этилового (этанол) автоматическими средствами измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола);
б) программно-аппаратные средства организаций, использующих фармацевтическую субстанцию спирта этилового (этанола) для производства спиртосодержащих лекарственных препаратов и в процессе производства других лекарственных средств, должны обеспечивать прием и передачу информации, полученной с использованием автоматических средств измерения и учета концентрации и объема безводного спирта в фармацевтической субстанции спирта этилового (этанола), объема фармацевтической субстанции спирта этилового (этанола), об объеме ее закупки;
в) оснащение оборудования для учета объема оборота и использования фармацевтической субстанции спирта этилового (этанола) для производства спиртосодержащих лекарственных препаратов, а также в процессе производства других лекарственных средств с использованием фармацевтической субстанции спирта этилового (этанола) техническими средствами фиксации и передачи информации об объеме производства и оборота спиртосодержащей продукции в единую государственную автоматизированную информационную систему. Требования настоящего подпункта не распространяются на лицензиатов, осуществляющих производство спиртосодержащих лекарственных препаратов, включенных в перечень спиртосодержащих лекарственных препаратов, утвержденный Правительством Российской Федерации в соответствии с абзацем третьим пункта 4 статьи 1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
г) программно-аппаратные средства организаций, использующих фармацевтическую субстанцию спирта этилового (этанола) для производства спиртосодержащих лекарственных препаратов и в процессе производства других лекарственных средств, должны обеспечивать прием и передачу информации об объеме использования фармацевтической субстанции спирта этилового (этанола), а также информации об объеме производства и (или) оборота (за исключением розничной продажи) спиртосодержащих лекарственных препаратов, полученной с применением технических средств фиксации и передачи информации об объеме производства и оборота спиртосодержащей продукции, в единую государственную автоматизированную информационную систему. Требования настоящего подпункта не распространяются на лицензиатов, осуществляющих производство спиртосодержащих лекарственных препаратов, включенных в перечень спиртосодержащих лекарственных препаратов, утвержденный Правительством Российской Федерации в соответствии с абзацем третьим пункта 4 статьи 1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
д) представление деклараций об объеме производства и (или) оборота (за исключением розничной продажи) спиртосодержащих лекарственных препаратов при производстве и (или) обороте спиртосодержащих лекарственных препаратов в объеме, превышающем 200 декалитров в год. Требования настоящего подпункта не распространяются на лицензиатов, осуществляющих производство спиртосодержащих лекарственных препаратов, включенных в перечень спиртосодержащих лекарственных препаратов, утвержденный Правительством Российской Федерации в соответствии с абзацем третьим пункта 4 статьи 1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
е) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" об использовании только фармацевтической субстанции спирта этилового (этанола) при производстве лекарственных средств в качестве действующего и (или) вспомогательного вещества, а также в иных технологических целях;
ж) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" о запрете производства спиртосодержащих лекарственных препаратов по месту осуществления производства фармацевтической субстанции спирта этилового (этанола) и (или) по месту осуществления производства этилового спирта;
з) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" о запрете производства спиртосодержащих лекарственных препаратов на основном технологическом оборудовании для производства этилового спирта, указанном в пункте 1.1 статьи 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции";
и) соблюдение лицензиатом требований статьи 45 Федерального закона "Об обращении лекарственных средств" об осуществлении перевозки фармацевтической субстанции спирта этилового (этанола) с соблюдением требований, установленных статьей 9 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции".
6. Осуществление деятельности по производству лекарственных средств с грубым нарушением лицензионных требований влечет за собой ответственность, установленную законодательством Российской Федерации.
При этом под грубым нарушением понимается невыполнение лицензиатом требований, предусмотренных подпунктами "а" - "и", "к(1)", "м" и "н" пункта 5 и пунктами 5(1) и 5(2) настоящего Положения, повлекшее за собой последствия, предусмотренные частью 10 статьи 19(2) Федерального закона "О лицензировании отдельных видов деятельности".
7. Для получения лицензии соискатель лицензии направляет или представляет в лицензирующий орган заявление, документы (копии документов) и сведения, указанные соответственно в части 1 и пунктах 1, 3 и 4 части 3 статьи 13 Федерального закона "О лицензировании отдельных видов деятельности", а также:
копии документов, подтверждающих наличие у соискателя лицензии на праве собственности или на ином законном основании необходимых для осуществления деятельности по производству лекарственных средств помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, права на которые не зарегистрированы в Едином государственном реестре прав на недвижимое имущество и сделок с ним (в случае если такие права зарегистрированы в указанном реестре - сведения об этих зданиях и помещениях), а также копии титульных листов промышленных регламентов;
копии документов, подтверждающих наличие в том числе соответствующие лицензионным требованиям образование, квалификацию и стаж работы уполномоченного лица производителя лекарственных средств, а также образование специалистов, ответственных за производство, маркировку и контроль качества лекарственных средств;
при намерении осуществлять производство фармацевтической субстанции спирта этилового (этанола) - копии документов, подтверждающих наличие на праве собственности, хозяйственного ведения или оперативного управления основного технологического оборудования для производства этилового спирта, зарегистрированного в соответствии со статьей 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции". Сведения о регистрации указанного оборудования представляются федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в области производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции, на основании межведомственного запроса лицензирующих органов.
8. При намерении осуществлять деятельность по производству лекарственных средств по адресу, не предусмотренному реестром лицензий, лицензиат в заявлении о внесении изменений в реестр лицензий в дополнение к сведениям, указанным в пунктах 1 и 3 части 1 статьи 13 и части 4.1 статьи 18 Федерального закона "О лицензировании отдельных видов деятельности", указывает:
а) сведения, содержащие новый адрес осуществления деятельности по производству лекарственных средств;
б) сведения о составляющих деятельность по производству лекарственных средств работах, которые лицензиат намерен выполнять по новому адресу осуществления деятельности по производству лекарственных средств;
в) сведения о наличии у лицензиата на праве собственности или на ином законном основании необходимых для осуществления деятельности по производству лекарственных средств по указанному новому адресу помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, а также промышленных регламентов;
г) сведения о наличии работников, заключивших трудовые договоры, имеющих соответствующее высшее или среднее профессиональное образование, ответственных за производство, маркировку и контроль качества лекарственных средств, намеренных осуществлять деятельность по указанному новому адресу;
д) при намерении осуществлять производство фармацевтической субстанции спирта этилового (этанола) по указанному новому адресу - копии документов, подтверждающих наличие на праве собственности, хозяйственного ведения или оперативного управления основного технологического оборудования для производства этилового спирта, зарегистрированного в соответствии со статьей 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции". Сведения о регистрации указанного оборудования представляются федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в области производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции, на основании межведомственного запроса лицензирующих органов.
9. При намерении осуществлять новые работы, составляющие деятельность по производству лекарственных средств, ранее не предусмотренные реестром лицензий, лицензиат в заявлении о внесении изменений в реестр лицензий в дополнение к сведениям, указанным в пунктах 1 и 3 части 1 статьи 13 Федерального закона "О лицензировании отдельных видов деятельности", указывает:
а) сведения о составляющих деятельность по производству лекарственных средств новых работах, которые лицензиат намерен выполнять;
б) сведения о наличии у лицензиата на праве собственности или на ином законном основании необходимых для осуществления новых работ, составляющих деятельность по производству лекарственных средств, помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, а также промышленных регламентов;
в) сведения о наличии работников, заключивших трудовые договоры, имеющих соответствующее высшее или среднее профессиональное образование, ответственных за производство, маркировку и контроль качества лекарственных средств, намеренных осуществлять новые работы, составляющие деятельность по производству лекарственных средств;
г) при намерении осуществлять производство фармацевтической субстанции спирта этилового (этанола) - копии документов, подтверждающих наличие на праве собственности, хозяйственного ведения или оперативного управления основного технологического оборудования для производства этилового спирта, зарегистрированного в соответствии со статьей 14.1 Федерального закона "О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции". Сведения о регистрации указанного оборудования представляются федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в области производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции, на основании межведомственного запроса лицензирующих органов.
10. При проведении проверки соответствия сведений, содержащихся в представленных соискателем лицензии (лицензиатом) заявлении и прилагаемых к нему документах, лицензионным требованиям лицензирующий орган запрашивает необходимые сведения у органов, предоставляющих государственные услуги, органов, предоставляющих муниципальные услуги, иных государственных органов, органов местного самоуправления либо подведомственных им организаций в порядке, установленном Федеральным законом "Об организации предоставления государственных и муниципальных услуг".
11. Лицензирующий орган размещает в федеральной государственной информационной системе "Единый портал государственных и муниципальных услуг (функций)" в установленном порядке сведения о ходе принятия им решения о предоставлении лицензии (об отказе в предоставлении лицензии), внесении изменений в реестр лицензий (об отказе во внесении изменений в реестр лицензий), приостановлении, возобновлении, прекращении действия лицензии, сведения об аннулировании лицензии, сведения о ходе принятия им решения о периодическом подтверждении соответствия лицензиата лицензионным требованиям, а также сведения о дате предоставления лицензии и регистрационном номере лицензии.
12. Доступ к общедоступной информации, содержащейся в реестрах лицензий, обеспечивается лицензирующим органом посредством ее размещения в информационно-телекоммуникационной сети "Интернет", в том числе в форме открытых данных. Данные о лицензиях, содержащиеся в реестре лицензий, получают статус открытых данных при внесении записи в этот реестр, который ведется в электронном виде.
13. Уполномоченные должностные лица лицензирующего органа при осуществлении лицензирования для установления соответствия соискателя лицензии или лицензиата лицензионным требованиям проводят оценку соответствия соискателя лицензии или лицензиата лицензионным требованиям в соответствии со статьей 19.1 Федерального закона "О лицензировании отдельных видов деятельности" в форме документарной и (или) выездной оценки, в том числе с использованием средств дистанционного взаимодействия.
Документарная оценка проводится при оценке сведений, содержащихся в представленных заявлениях и документах, в целях оценки соответствия таких сведений положениям частей 1 и 3 статьи 13 и части 3 статьи 18 Федерального закона "О лицензировании отдельных видов деятельности", а также сведений о соискателе лицензии или лицензиате, содержащихся в Едином государственном реестре юридических лиц и других федеральных информационных ресурсах.
Кроме документарной оценки, проводится выездная оценка в отношении соискателя лицензии при предоставлении лицензии и лицензиата в случае внесения изменений в реестр лицензий при намерении лицензиата осуществлять деятельность по производству лекарственных средств по новому адресу, не предусмотренному реестром лицензий, и в случае внесения изменений в реестр лицензий при намерении лицензиата выполнять новые работы, составляющие деятельность по производству лекарственных средств, ранее не предусмотренные реестром лицензий.
Выездная оценка проводится в целях оценки соответствия лицензионным требованиям состояния производственных объектов, технических средств, оборудования, иных объектов, которые предполагается использовать соискателем лицензии или лицензиатом при осуществлении производства лекарственных средств и которые необходимы для осуществления деятельности по производству лекарственных средств.
Выездная оценка с использованием средств дистанционного взаимодействия проводится в случаях, определенных приложением N 2.1 к Правилам проведения фармацевтических инспекций, утвержденным Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 83 "Об утверждении Правил проведения фармацевтических инспекций".
При проведении оценки соответствия соискателя лицензии и лицензиата лицензионным требованиям, предъявляемым к соискателю лицензии и лицензиату на осуществление деятельности по производству лекарственных средств для медицинского применения, лицензирующий орган вправе привлекать федеральное бюджетное учреждение "Государственный институт лекарственных средств и надлежащих практик", подведомственное Министерству промышленности и торговли Российской Федерации.
При проведении оценки соответствия соискателя лицензии и лицензиата лицензионным требованиям, предъявляемым к соискателю лицензии и лицензиату на осуществление деятельности по производству лекарственных средств для ветеринарного применения, лицензирующий орган вправе привлекать федеральное государственное бюджетное учреждение "Всероссийский государственный Центр качества и стандартизации лекарственных средств для животных и кормов", подведомственное Федеральной службе по ветеринарному и фитосанитарному надзору.
Формирование государственного задания на участие в проведении оценки соответствия соискателя лицензии и лицензиата лицензионным требованиям в отношении федерального бюджетного учреждения "Государственный институт лекарственных средств и надлежащих практик" и федерального государственного бюджетного учреждения "Всероссийский государственный Центр качества и стандартизации лекарственных средств для животных и кормов" и финансовое обеспечение выполнения указанного государственного задания осуществляются в порядке, установленном постановлением Правительства Российской Федерации от 26 июня 2015 г. N 640 "О порядке формирования государственного задания на оказание государственных услуг (выполнение работ) в отношении федеральных государственных учреждений и финансового обеспечения выполнения государственного задания".
13(1). Обеспечение соблюдения лицензиатом лицензионных требований осуществляется путем проведения периодического подтверждения соответствия лицензиатов лицензионным требованиям, осуществляемого в форме государственной услуги.
Периодическое подтверждение соответствия лицензиатов лицензионным требованиям проводится в соответствии со статьей 19.3 Федерального закона "О лицензировании отдельных видов деятельности".
При проведении периодического подтверждения соответствия лицензиатов лицензионным требованиям могут привлекаться подведомственные лицензирующему органу организации, указанные в пункте 13 настоящего Положения, путем формирования государственного задания в соответствии с пунктом 13 настоящего Положения.
14. Представление соискателем лицензии заявления и документов, необходимых для получения лицензии, и их прием лицензирующим органом, принятие лицензирующим органом решения о предоставлении лицензии (об отказе в предоставлении лицензии), внесении изменений в реестр лицензий (об отказе во внесении изменений в реестр лицензий), приостановлении, возобновлении, прекращении действия лицензии, а также предоставление выписки из реестра лицензий, формирование и ведение лицензионного дела, формирование и ведение реестра лицензий и предоставление сведений, содержащихся в реестре лицензий, осуществляются в порядке, установленном Федеральным законом "О лицензировании отдельных видов деятельности".
15. За предоставление лицензирующим органом лицензии и внесение изменений в реестр лицензий на основании заявления лицензиата о внесении изменений в реестр лицензий, поданного в лицензирующий орган в соответствии со статьями 13 и 18 Федерального закона "О лицензировании отдельных видов деятельности", уплачивается государственная пошлина в размерах и порядке, которые установлены законодательством Российской Федерации о налогах и сборах.
16. Федеральный государственный лицензионный контроль деятельности по производству лекарственных средств осуществляется в соответствии с Положением о федеральном государственном лицензионном контроле деятельности по производству лекарственных средств согласно приложению N 2.
Приложение N 1
к Положению о лицензировании
производства лекарственных средств
ПЕРЕЧЕНЬ
РАБОТ, СОСТАВЛЯЮЩИХ ДЕЯТЕЛЬНОСТЬ ПО ПРОИЗВОДСТВУ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
I. В сфере обращения лекарственных средств
для медицинского применения <1>
Производство лекарственных средств для медицинского применения и (или) лекарственных препаратов для клинических исследований (испытаний):
1. Производственные операции - лекарственная продукция <2>.
1.1. Стерильная продукция:
1.1.1. Производимая асептическим путем (операции обработки для следующих лекарственных форм):
1.1.1.1. Жидкие лекарственные формы большого объема.
1.1.1.2. Жидкие лекарственные формы малого объема.
1.1.1.3. Лиофилизаты.
1.1.1.4. Твердые лекарственные формы и имплантаты.
1.1.1.5. Мягкие лекарственные формы.
1.1.1.6. Прочая продукция.
1.1.2. Подвергаемая финишной стерилизации (операции обработки для следующих лекарственных форм):
1.1.2.1. Жидкие лекарственные формы большого объема.
1.1.2.2. Жидкие лекарственные формы малого объема.
1.1.2.3. Твердые лекарственные формы и имплантаты.
1.1.2.4. Мягкие лекарственные формы.
1.1.2.5. Прочая продукция, лекарственные формы.
1.1.3. Выпускающий контроль (сертификация серий).
1.2. Нестерильная продукция:
1.2.1. Нестерильная продукция (операции обработки для следующих лекарственных форм):
1.2.1.1. Капсулы в твердой оболочке.
1.2.1.2. Капсулы в мягкой оболочке.
1.2.1.3. Жевательные лекарственные формы.
1.2.1.4. Импрегнированные лекарственные формы.
1.2.1.5. Жидкие лекарственные формы для наружного применения.
1.2.1.6. Жидкие лекарственные формы для внутреннего применения.
1.2.1.7. Медицинские газы.
1.2.1.8. Прочие твердые лекарственные формы.
1.2.1.9. Препараты, находящиеся под давлением.
1.2.1.10. Радионуклидные генераторы.
1.2.1.11. Мягкие лекарственные формы.
1.2.1.12. Свечи (суппозитории).
1.2.1.13. Таблетки.
1.2.1.14. Трансдермальные пластыри.
1.2.1.15. Прочая продукция, лекарственные формы.
1.2.2. Выпускающий контроль (сертификация серий).
1.3. Биологическая лекарственная продукция:
1.3.1. Биологическая лекарственная продукция <3>:
1.3.1.1. Продукты крови.
1.3.1.2. Иммунологическая (иммунобиологическая) продукция.
1.3.1.3. Продукты на основе соматических клеток.
1.3.1.4. Генотерапевтическая продукция.
1.3.1.5. Биотехнологическая продукция.
1.3.1.6. Продукты, экстрагированные из животных источников или органов (тканей) человека.
1.3.1.7. Продукты тканевой инженерии.
1.3.1.8. Прочая биологическая продукция.
1.3.2. Выпускающий контроль (сертификация серий) (перечень видов продукции):
1.3.2.1. Продукты крови.
1.3.2.2. Иммунологическая (иммунобиологическая) продукция.
1.3.2.3. Продукты на основе соматических клеток.
1.3.2.4. Генотерапевтическая продукция.
1.3.2.5. Биотехнологическая продукция.
1.3.2.6. Продукты, экстрагированные из животных источников или органов (тканей) человека.
1.3.2.7. Продукты тканевой инженерии.
1.3.2.8. Прочая биологическая продукция.
1.4. Прочая лекарственная продукция или производственные операции:
1.4.1. Производство <4>:
1.4.1.1. Растительная продукция.
1.4.1.2. Гомеопатическая продукция.
1.4.1.3. Прочая продукция.
1.4.2. Стерилизация фармацевтических субстанций, вспомогательных веществ, готовой продукции:
1.4.2.1. Фильтрация.
1.4.2.2. Сухожаровая стерилизация.
1.4.2.3. Стерилизация паром.
1.4.2.4. Химическая стерилизация.
1.4.2.5. Стерилизация гамма-излучением.
1.4.2.6. Стерилизация электронным излучением.
1.4.3. Прочее.
1.5. Упаковка:
1.5.1. Первичная упаковка:
1.5.1.1. Капсулы в твердой оболочке.
1.5.1.2. Капсулы в мягкой оболочке.
1.5.1.3. Жевательные лекарственные формы.
1.5.1.4. Импрегнированные лекарственные формы.
1.5.1.5. Жидкие лекарственные формы для наружного применения.
1.5.1.6. Жидкие лекарственные формы для внутреннего применения.
1.5.1.7. Медицинские газы.
1.5.1.8. Прочие твердые лекарственные формы.
1.5.1.9. Препараты, находящиеся под давлением.
1.5.1.10. Радионуклидные генераторы.
1.5.1.11. Мягкие лекарственные формы.
1.5.1.12. Свечи (суппозитории).
1.5.1.13. Таблетки.
1.5.1.14. Трансдермальные пластыри.
1.5.1.15. Прочая продукция.
1.5.2. Вторичная упаковка.
1.6. Испытания контроля качества:
1.6.1. Микробиологические: стерильность.
1.6.2. Микробиологические: микробиологическая чистота.
1.6.3. Химические (физические).
1.6.4. Биологические.
1.7. Хранение и реализация лекарственных средств:
1.7.1. Хранение произведенных лекарственных средств.
1.7.2. Реализация и (или) передача произведенных лекарственных средств.
1.8. Маркировка лекарственных средств, подлежащих обязательной маркировке средствами идентификации:
1.8.1. Сериализация.
1.8.2. Агрегация.
2. Импорт лекарственной продукции <2>.
2.1. Испытания контроля качества импортируемой лекарственной продукции <5>:
2.1.1. Микробиологические: стерильность.
2.1.2. Микробиологические: микробиологическая чистота.
2.1.3. Химические (физические).
2.1.4. Биологические.
2.2. Выпускающий контроль (сертификация серий) импортируемой лекарственной продукции <6>:
2.2.1. Стерильная продукция:
2.2.1.1. Производимая асептическим путем.
2.2.1.2. Подвергаемая финишной стерилизации.
2.2.2. Нестерильная продукция.
2.2.3. Биологическая лекарственная продукция:
2.2.3.1. Продукты крови.
2.2.3.2. Иммунологическая (иммунобиологическая) продукция.
2.2.3.3. Продукты на основе соматических клеток.
2.2.3.4. Генотерапевтическая продукция.
2.2.3.5. Биотехнологическая продукция.
2.2.3.6. Продукты, экстрагированные из животных источников или органов (тканей) человека.
2.2.3.7. Продукты тканевой инженерии.
2.2.3.8. Прочая биологическая продукция.
2.3. Прочая деятельность по импорту (ввозу) <7>:
2.3.1. Площадка физического импорта (ввоза).
2.3.2. Импорт промежуточного продукта, подвергающегося дальнейшей обработке.
2.3.3. Прочее.
2.4. Маркировка лекарственных средств, подлежащих обязательной маркировке средствами идентификации:
2.4.1. Сериализация.
2.4.2. Агрегация.
3. Производственные операции (фармацевтические субстанции) <2>.
3.1. Производство фармацевтических субстанций методом химического синтеза:
3.1.1. Производство промежуточных продуктов фармацевтической субстанции.
3.1.2. Производство неочищенной (необработанной) фармацевтической субстанции.
3.1.3. Завершающие стадии производственного процесса (например: очистка, обработка физическими методами).
3.1.4. Прочее.
3.2. Производство фармацевтических субстанций методом выделения из природных источников:
3.2.1. Выделение фармацевтических субстанций из источников растительного происхождения.
3.2.2. Выделение фармацевтических субстанций из источников животного происхождения.
3.2.3. Выделение фармацевтических субстанций из источников из органов (тканей) человека.
3.2.4. Выделение фармацевтических субстанций из источников минерального происхождения.
3.2.5. Модификация выделенной фармацевтической субстанции (указывается источник 3.2.1, 3.2.2, 3.2.3, 3.2.4).
3.2.6. Очистка выделенной фармацевтической субстанции (указывается источник 3.2.1, 3.2.2, 3.2.3, 3.2.4).
3.2.7. Прочее.
3.3. Производство фармацевтических субстанций с использованием биологических процессов:
3.3.1. Ферментация (брожение).
3.3.2. Производство с использованием клеточных культур (указывается тип используемых клеток).
3.3.3. Выделение (Очистка).
3.3.4. Модификация.
3.3.5. Прочее.
3.3(1). Производство фармацевтической субстанции спирта этилового (этанола):
3.3(1).1. Производство методом разведения водой очищенной ректификованного этилового спирта из пищевого сырья, произведенного по месту осуществления производства фармацевтической субстанции спирта этилового (этанола).
3.4. Производство стерильных фармацевтических субстанций (разделы 3.1, 3.2, 3.3 должны заполняться по мере их применимости):
3.4.1. Производимые в асептических условиях.
3.4.2. Подвергаемые финишной стерилизации.
3.5. Завершающие стадии производства фармацевтических субстанций:
3.5.1. Стадии физической обработки (указывается вид обработки, например: сушка, измельчение, просеивание).
3.5.2. Первичная упаковка.
3.5.3. Вторичная упаковка.
3.5.4. Прочее (для операций, не описанных выше).
3.6. Испытания контроля качества:
3.6.1. Химические (физические).
3.6.2. Микробиологические: микробиологическая чистота.
3.6.3. Микробиологические: стерильность.
3.6.4. Биологические.
3.7. Хранение и реализация фармацевтических субстанций:
3.7.1.1. Хранение произведенных фармацевтических субстанций.
3.7.1.2. Реализация и (или) передача произведенных фармацевтических субстанций.
II. В сфере обращения лекарственных средств
для ветеринарного применения
4. Производство, хранение и реализация фармацевтических субстанций, получаемых методами химического синтеза.
5. Производство, хранение и реализация фармацевтических субстанций, получаемых методами биотехнологического синтеза.
6. Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из химического сырья.
7. Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из источников биологического, животного происхождения.
8. Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из источников растительного происхождения.
8.1. Производство, хранение и реализация фармацевтической субстанции спирта этилового (этанола).
9. Производство, хранение и реализация стерильных лекарственных препаратов с указанием конкретной лекарственной формы (аэрозоль, гель, крем, линимент, лиофилизированные продукты, мазь, паста, пленка, порошок, раствор, раствор для инъекций, стеклообразная масса, стерильная пористая масса, суспензия, сухая масса, таблетки, эмульсии).
10. Производство, хранение и реализация нестерильных лекарственных препаратов с указанием конкретной лекарственной формы (аэрозоль, бальзам, брикет, гель, гранулы, драже, капли, капсулы, капсулы мягкие, крем, линимент, мазь, масло, микрогранулы, микрокапсулы, настой, настойка, пастилки, паста, пеллеты, пластины, пластинки, пластырь, пленка, полимерная лента, полоски, порошок, раствор, сироп, спрей, суппозитории, суспензия, таблетки, шнур, экстракт, эликсир, эмульсия).
--------------------------------
<1> Лекарственные формы указываются в соответствии с номенклатурой лекарственных форм, утвержденной Евразийской экономической комиссией. В случае отсутствия в номенклатуре лекарственных форм, утвержденной Евразийской экономической комиссией, лекарственной формы, заявленной производителем, такая лекарственная форма указывается в соответствии с перечнем наименований лекарственных форм, утвержденным Министерством здравоохранения Российской Федерации.
<2> Указываются применимые пункты.
<3> В категории 1.3.1. "Биологическая лекарственная продукция" в соответствующем подразделе обязательно указываются группы препаратов (аллергены, аллергоиды, анатоксины, вакцины, гаммаглобулины, иммуноглобулины, иммуномодуляторы, моноклональные антитела, сыворотки, токсины, цитокины, препараты, получаемые из животного сырья, инсулины, органопрепараты, бактериофаги, вакцины, иммуноглобулиновые комплексные препараты, пробиотики, прочая биотехнологическая продукция).
<4> В категории 1.4.1. "Производство" в подразделе 1.4.1.3 обязательно указываются группы препаратов (радиофармацевтические лекарственные средства, цитостатики, цитотоксики, гормоны, антибиотики бета-лактамного ряда, препараты, содержащие сильнодействующие вещества, наркотические лекарственные средства, психотропные лекарственные средства).
<5> В категории 2.1. "Испытания контроля качества импортируемой лекарственной продукции" указывается соответствующий подраздел в случае осуществления соответствующего испытания контроля качества на производственном объекте в отношении импортируемых лекарственных средств.
<6> В категории 2.2. "Выпускающий контроль (сертификация серий) импортируемой лекарственной продукции" указывается в случае осуществления подтверждения соответствия уполномоченным лицом производителя в отношении импортируемых лекарственных средств.
<7> В категории 2.3. "Прочая деятельность по импорту (ввозу)" подраздел 2.3.1 указывается только в случае хранения импортируемых лекарственных средств, ожидающих подтверждения соответствия уполномоченным лицом производителя.
Приложение N 2
к Положению о лицензировании
производства лекарственных средств
ПОЛОЖЕНИЕ
О ФЕДЕРАЛЬНОМ ГОСУДАРСТВЕННОМ ЛИЦЕНЗИОННОМ КОНТРОЛЕ
ДЕЯТЕЛЬНОСТИ ПО ПРОИЗВОДСТВУ ЛЕКАРСТВЕННЫХ СРЕДСТВ
I. Общие положения
1. Настоящее Положение устанавливает порядок организации и осуществления федерального государственного лицензионного контроля деятельности по производству лекарственных средств (далее - лицензионный контроль).
2. Лицензионный контроль в отношении деятельности по производству лекарственных средств для медицинского применения осуществляет Министерство промышленности и торговли Российской Федерации, в отношении деятельности по производству лекарственных средств для ветеринарного применения - Федеральная служба по ветеринарному и фитосанитарному надзору (далее - лицензирующий орган) в соответствии с Федеральным законом "О лицензировании отдельных видов деятельности", Федеральным законом "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации" и настоящим Положением.
3. Предметом лицензионного контроля является соблюдение лицензиатами (контролируемыми лицами) лицензионных требований в отношении деятельности по производству лекарственных средств, установленных Положением о лицензировании производства лекарственных средств, утвержденным постановлением Правительства Российской Федерации от 6 июля 2012 г. N 686 "Об утверждении Положения о лицензировании производства лекарственных средств" (далее - деятельность по производству лекарственных средств). Под контролируемым лицом понимается лицензиат, осуществляющий деятельность по производству лекарственных средств.
Обеспечение соблюдения лицензиатом лицензионных требований осуществляется путем проведения профилактических мероприятий и внеплановых контрольных (надзорных) мероприятий (далее - контрольное мероприятие) в соответствии с Федеральным законом "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации".
4. Объектами лицензионного контроля являются:
деятельность лицензиатов по производству лекарственных средств для медицинского применения;
деятельность лицензиатов по производству лекарственных средств для ветеринарного применения.
5. Учет объектов лицензионного контроля осуществляется посредством сбора, обработки, анализа и учета сведений об объектах лицензионного контроля, информации, представляемой лицензирующему органу в соответствии с нормативными правовыми актами Российской Федерации, информации, получаемой в рамках межведомственного взаимодействия, а также общедоступной информации без взаимодействия с контролируемыми лицами.
6. Учет объектов лицензионного контроля осуществляется лицензирующим органом посредством ведения перечня объектов лицензионного контроля. Перечень объектов лицензионного контроля содержит следующую информацию:
полное наименование юридического лица;
идентификационный номер налогоплательщика юридического лица;
адрес места нахождения и осуществления деятельности контролируемых лиц по производству лекарственных средств и используемых ими производственных объектов;
вид (виды) деятельности в соответствии с Общероссийским классификатором видов экономической деятельности.
Размещение информации, указанной в абзацах втором - пятом настоящего пункта, осуществляется с учетом требований законодательства Российской Федерации о государственной тайне.
II. Должностные лица, уполномоченные
на принятие решения о проведении контрольного мероприятия
и осуществление лицензионного контроля
7. Должностными лицами, уполномоченными на принятие решений о проведении контрольного мероприятия, являются руководители лицензирующих органов или иные должностные лица лицензирующих органов, на которых осуществление указанного полномочия возложено в соответствии с приказом Министерства промышленности и торговли Российской Федерации или приказом Федеральной службы по ветеринарному и фитосанитарному надзору.
8. Должностными лицами, уполномоченными на осуществление лицензионного контроля, являются руководители лицензирующих органов или должностные лица, должности которых соответствуют следующим группам должностей:
высшие должности гражданской службы;
главные должности гражданской службы;
ведущие должности гражданской службы;
старшие должности гражданской службы;
младшие должности гражданской службы Министерства промышленности и торговли Российской Федерации или Федеральной службы по ветеринарному и фитосанитарному надзору, на которых в соответствии с должностными регламентами возложено осуществление лицензионного контроля.
III. Профилактические мероприятия в рамках осуществления
лицензионного контроля
9. В целях профилактики рисков причинения вреда жизни, здоровью граждан и животных в рамках лицензионного контроля лицензирующим органом ежегодно утверждается программа профилактики рисков причинения вреда жизни, здоровью граждан и животным в соответствии с Правилами разработки и утверждения контрольными (надзорными) органами программы профилактики рисков причинения вреда (ущерба) охраняемым законом ценностям, утвержденными постановлением Правительства Российской Федерации от 25 июня 2021 г. N 990 "Об утверждении Правил разработки и утверждения контрольными (надзорными) органами программы профилактики рисков причинения вреда (ущерба) охраняемым законом ценностям".
10. Уполномоченные должностные лица лицензирующего органа обязаны проводить следующие виды профилактических мероприятий:
информирование;
обобщение правоприменительной практики;
объявление предостережения;
консультирование;
профилактический визит.
11. Информирование контролируемых лиц и иных заинтересованных лиц по вопросам соблюдения обязательных требований в отношении деятельности по производству лекарственных средств осуществляется лицензирующим органом посредством размещения соответствующих сведений на своем официальном сайте в информационно-телекоммуникационной сети "Интернет" (далее - сеть "Интернет"), в средствах массовой информации, через личные кабинеты контролируемых лиц в государственных информационных системах (при их наличии) в течение 5 рабочих дней со дня утверждения нормативного правового акта Российской Федерации, содержащего обязательные требования.
12. Обобщение правоприменительной практики оформляется в виде доклада лицензирующим органом ежегодно.
Проект доклада размещается на официальном сайте лицензирующего органа в сети "Интернет" для прохождения процедуры его публичного обсуждения. Срок публичного обсуждения проекта доклада - 1 месяц со дня его размещения на указанном сайте.
Доклад утверждается приказом руководителя лицензирующего органа и размещается на официальном сайте лицензирующего органа в сети "Интернет" до 15 марта года, следующего за отчетным годом.
13. В случае наличия у лицензирующего органа сведений о готовящихся нарушениях обязательных требований или признаках нарушений обязательных требований в сфере производства лекарственных средств и (или) в случае отсутствия подтвержденных данных о том, что нарушение обязательных требований причинило вред (ущерб) охраняемым законом ценностям или создало угрозу причинения вреда (ущерба) охраняемым законом ценностям, лицензирующий орган объявляет контролируемому лицу предостережение о недопустимости нарушения обязательных требований и предлагает принять меры по обеспечению соблюдения обязательных требований (далее - предостережение).
Решение о направлении предостережения принимает руководитель, заместитель руководителя лицензирующего органа или иное уполномоченное приказом лицензирующего органа должностное лицо лицензирующего органа на основании предложений должностного лица лицензирующего органа при наличии сведений, указанных в абзаце первом настоящего пункта.
Предостережение направляется на бумажном носителе заказным почтовым отправлением с уведомлением о вручении или иным доступным для контролируемого лица способом, включая направление в виде электронного документа, подписанного усиленной квалифицированной электронной подписью лица, принявшего решение об объявлении предостережения, с использованием сети "Интернет", в том числе по адресу электронной почты контролируемого лица, указанному соответственно в Едином государственном реестре юридических лиц или размещенному на официальном сайте контролируемого лица в составе информации, размещение которой является обязательным в соответствии с законодательством Российской Федерации, или посредством федеральной государственной информационной системы "Единый портал государственных и муниципальных услуг (функций)".
По результатам рассмотрения предостережения контролируемое лицо вправе подать возражение в лицензирующий орган, направивший предостережение.
Возражения направляются контролируемым лицом на бумажном носителе почтовым отправлением в лицензирующий орган, или в виде электронного документа, подписанного электронной подписью уполномоченного лица производителя лекарственных средств, на указанный в предостережении адрес электронной почты лицензирующего органа, или иными указанными в предостережении способами в течение 30 календарных дней со дня его получения.
Лицензирующий орган по итогам рассмотрения возражения направляет контролируемому лицу в течение 30 рабочих дней со дня получения возражений результаты рассмотрения возражений.
Лицензирующий орган осуществляет учет объявленных им предостережений и использует соответствующие данные для проведения иных профилактических и контрольных мероприятий.
14. Консультирование контролируемых лиц осуществляется лицензирующим органом в письменной форме при их письменном обращении, в устной форме по телефону, посредством видео-конференц-связи или на личном приеме у уполномоченного лица.
15. Консультирование контролируемых лиц в ходе проведения профилактического мероприятия, контрольного мероприятия осуществляется до завершения соответствующего мероприятия в случае волеизъявления контролируемого лица, о чем делается отметка в документах, оформляемых по итогам соответствующего мероприятия.
16. Должностные лица лицензирующего органа лицензионного контроля осуществляют консультирование контролируемых лиц по следующим вопросам:
наличие и (или) содержание обязательных требований в отношении деятельности по производству лекарственных средств;
периодичность и порядок проведения контрольных мероприятий;
порядок выполнения обязательных требований при осуществлении деятельности по производству лекарственных средств;
выполнение предписания, выданного по итогам контрольного мероприятия.
17. Должностные лица лицензирующего органа осуществляют письменное консультирование контролируемых лиц по вопросам, предусмотренным абзацем пятым пункта 16 настоящего Положения.
18. В ходе консультирования не могут предоставляться информация, содержащая оценку конкретного контрольного мероприятия, решений и (или) действий должностных лиц лицензирующего органа, иных участников контрольного мероприятия, а также результаты проведенных в рамках контрольного мероприятия экспертиз.
19. Консультирование контролируемых лиц по однотипным обращениям контролируемых лиц и их представителей посредством размещения на официальном сайте лицензирующего органа в сети "Интернет" письменного разъяснения, подписанного уполномоченным должностным лицом лицензирующего органа, осуществляется в случаях регулярного поступления обращений (более 5) по вопросу соблюдения одних и тех же обязательных требований.
20. Обязательные профилактические визиты проводятся в отношении контролируемых лиц, приступивших в течение одного года, предшествующего принятию решения о проведении профилактического визита, к осуществлению деятельности в сфере производства лекарственных средств (контролируемых лиц, получивших лицензии, контролируемых лиц, в отношении которых внесены изменения в реестр лицензий в связи с осуществлением не предусмотренных в ранее действовавшей лицензии работ и услуг, составляющих деятельность по производству лекарственных средств, и (или) осуществлением деятельности по производству лекарственных средств по адресу, не указанному в реестре лицензий).
Обязательный профилактический визит проводится в соответствии со статьей 52 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации" в рабочее время путем использования видео-конференц-связи в период, устанавливаемый уведомлением о проведении обязательного профилактического визита, и не может превышать 2 часа. При проведении обязательного профилактического визита должностными лицами лицензирующего органа осуществляется информирование контролируемого лица об обязательных требованиях, предъявляемых к осуществляемой им деятельности по производству лекарственных средств либо принадлежащим ему объектам лицензионного контроля, а также о видах, содержании и интенсивности контрольных мероприятий, проводимых в отношении объекта лицензионного контроля.
21. Целями проведения профилактических визитов являются:
а) предупреждение и сокращение количества нарушений контролируемыми лицами обязательных требований;
б) создание мотивации у контролируемых лиц к добросовестному поведению и, как следствие, снижение уровня ущерба охраняемым законом ценностям;
в) формирование единого понимания обязательных требований;
г) выявление причин, факторов и условий, способствующих нарушению обязательных требований, определение способов устранения или снижения рисков их возникновения.
22. По итогам завершения профилактического визита должностное лицо лицензирующего органа составляет акт проведения профилактического визита в 2 экземплярах, в котором указываются:
а) наименование лицензирующего органа;
б) наименование контролируемого лица;
в) дата, время и место составления акта профилактического визита;
г) реквизиты приказа, на основании которого проводился профилактический визит;
д) фамилии, имена, отчества (при наличии), наименования должностей должностных лиц лицензирующего органа, проводивших профилактический визит;
е) дата, время, продолжительность и место проведения профилактического визита;
ж) перечень мероприятий, проведенных в ходе профилактического визита;
з) сведения о результатах проведения профилактического визита;
и) перечень прилагаемых документов и материалов (при наличии);
к) подписи должностных лиц лицензирующего органа, проводивших профилактический визит.
IV. Осуществление государственного лицензионного контроля
23. Лицензионный контроль осуществляется посредством проведения внеплановых контрольных мероприятий.
При проведении лицензионного контроля могут привлекаться не заинтересованные в результатах такой оценки специалисты в соответствии со статьей 34 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации".
24. Внеплановые контрольные мероприятия во взаимодействии с проверяемыми контролируемыми лицами осуществляются в следующих видах контрольных мероприятий:
а) документарная проверка;
б) выездная проверка.
25. Внеплановые контрольные мероприятия, указанные в пункте 24 настоящего Положения, проводятся в соответствии со статьями 57, 66, 72 и 73 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации".
26. В ходе документарной проверки в соответствии со статьями 72, 79 и 80 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации" могут совершаться следующие контрольные действия:
а) получение письменных объяснений;
б) истребование документов.
27. В ходе выездной проверки в соответствии со статьями 73, 76, 78 - 81, 83 и 84 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации" могут совершаться следующие контрольные действия:
а) осмотр;
б) опрос;
в) получение письменных объяснений;
г) истребование документов;
д) отбор проб и образцов;
е) испытание;
ж) экспертиза.
28. Срок проведения выездной проверки устанавливается в пределах 10 рабочих дней.
Срок проведения выездной проверки в отношении контролируемого лица, осуществляющего свою деятельность на территориях нескольких субъектов Российской Федерации, устанавливается отдельно по каждому филиалу, представительству, обособленному структурному подразделению организации или производственному объекту.
29. В случае выявления при проведении внепланового контрольного мероприятия нарушений лицензионных требований в отношении деятельности по производству лекарственных средств, установленных Положением о лицензировании производства лекарственных средств, утвержденным постановлением Правительства Российской Федерации от 6 июля 2012 г. N 686 "Об утверждении Положения о лицензировании производства лекарственных средств", лицензирующий орган в пределах полномочий обязан принять решения, предусмотренные частью 2 статьи 90 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации".
30. В случае отсутствия контролируемого лица или его представителя отбор проб (образцов) продукции (товаров) осуществляется с применением видеозаписи.
31. В целях проведения испытаний и экспертизы лекарственных средств на соответствие требованиям нормативной документации (нормативного документа по качеству) лицензирующий орган привлекает специалиста и (или) аккредитованные в соответствии с законодательством Российской Федерации об аккредитации в национальной системе аккредитации экспертную организацию или федеральные государственные бюджетные учреждения, подведомственные лицензирующему органу (далее - экспертная организация).
32. Отбор проб (образцов) лекарственных средств осуществляется должностными лицами лицензирующего органа (территориального органа) в соответствии со своими должностными полномочиями.
33. Отбор проб (образцов) лекарственных средств для проведения испытаний экспертизы по всем или отдельным показателям нормативной документации (нормативного документа) осуществляется в количествах, необходимых для двукратного воспроизведения методов контроля качества лекарственного средства с учетом требований нормативной документации (нормативного документа) на него.
34. Для проведения испытаний и экспертизы методами неразрушающего анализа отбираются 3 упаковки лекарственного препарата или 10 граммов фармацевтической субстанции.
35. При совпадении спектров отобранных проб (образцов) лекарственных средств с эталонным спектром, установленным в соответствии с фармакопейными требованиями, отобранные пробы (образцы) лекарственных средств возвращаются контролируемому лицу, у которого они были отобраны для проведения экспертизы, с оформлением акта возврата образцов, форма которого утверждается лицензирующим органом.
36. При несовпадении спектра лекарственного средства с эталонным спектром осуществляется отбор проб (образцов) лекарственного средства для проведения экспертизы по показателям нормативной документации (нормативного документа) по качеству в соответствии с пунктами 37 и 38 настоящего Положения.
37. Пробы (образцы) лекарственных средств направляются лицензирующим органом (территориальным органом) в экспертную организацию с приложением копий протокола отбора проб (образцов) лекарственных средств.
38. Экспертиза осуществляется экспертом или экспертной организацией по поручению лицензирующего органа (территориального органа) на основании экспертного задания, которое может включать одну или несколько из следующих задач экспертизы:
а) установление фактов, обстоятельств;
б) установление тождества или различия;
в) установление объективных свойств и состояний имеющихся в наличии образцов лекарственных средств;
г) проведение оценки образца лекарственного средства на соответствие заданным критериям;
д) установление соответствия образца лекарственного средства требованиям нормативной документации (нормативного документа).
39. Время осуществления экспертизы зависит от вида экспертизы и устанавливается индивидуально в каждом конкретном случае по соглашению между лицензирующим органом и экспертом или экспертной организацией.
40. Фотосъемка, аудио-, видеозапись осуществляются в следующем порядке:
а) для фиксации хода и результатов контрольного мероприятия осуществляются ориентирующая, обзорная, узловая и детальная фотосъемка и видеозапись;
б) фото-, аудио-, видеофиксация проводятся должностным лицом, уполномоченным на осуществление лицензионного контроля, при проведении контрольного мероприятия посредством использования фотоаппаратов, диктофонов, видеокамер, а также мобильных устройств (телефонов, смартфонов, планшетов);
в) оборудование, используемое для проведения фото- и видеофиксации, должно иметь техническую возможность отображения на фотоснимках и видеозаписи текущей даты и времени, а также сохранения данных о месте съемки (координат);
г) аудиозапись ведет должностное лицо, уполномоченное на осуществление лицензионного контроля, при проведении контрольного мероприятия;
д) при проведении фото- и видеофиксации:
необходимо применять приемы фиксации, при которых исключается возможность искажения свойств объекта контроля;
следует обеспечивать условия фиксации, при которых полученные фотоснимки, видеозапись максимально точно и полно отображают свойства объектов контроля;
е) информация о проведении фотосъемки, аудио- и видеозаписи отражается в акте контрольного мероприятия с указанием типа и марки оборудования, с помощью которого проводилась фиксация;
ж) фото-, аудио- и видеоматериалы являются приложением к акту контрольного мероприятия.
V. Ключевые показатели лицензионного контроля
и их целевые значения
41. Ключевые показатели лицензионного контроля (КП) отражают уровень минимизации поступления недоброкачественных лекарственных средств в гражданский оборот.
Целевое значение ключевого показателя лицензионного контроля определяется исходя из ежегодного снижения на 1 процент.
Ключевой показатель лицензионного контроля (КП) определяется по формуле:
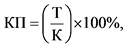
где:
Т - количество отозванных серий лекарственных препаратов в связи с нарушением требований к производству лекарственных средств за отчетный период;
К - общее количество серий лекарственных препаратов, находящихся в обращении на территории Российской Федерации за отчетный период.
VI. Обжалование решений контрольных (надзорных) органов,
действий (бездействия) их должностных лиц
42. Правом на обжалование решений Министерства промышленности и торговли Российской Федерации или Федеральной службы по ветеринарному и фитосанитарному надзору и ее территориальных органов, действий (бездействия) их должностных лиц обладает контролируемое лицо, в отношении которого приняты решения или совершены действия (бездействие), указанные в части 4 статьи 40 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации".
43. Жалоба на решение Министерства промышленности и торговли Российской Федерации, действия (бездействие) его должностных лиц рассматривается руководителем Министерства промышленности и торговли Российской Федерации.
Жалоба на решение территориального органа Федеральной службы по ветеринарному и фитосанитарному надзору, действия (бездействие) его должностных лиц рассматривается руководителем (заместителем руководителя) указанного территориального органа Федеральной службы по ветеринарному и фитосанитарному надзору.
Жалоба на действия (бездействие) руководителя (заместителя руководителя) территориального органа Федеральной службы по ветеринарному и фитосанитарному надзору рассматривается Федеральной службой по ветеринарному и фитосанитарному надзору.
В случае обжалования решений центрального аппарата Федеральной службы по ветеринарному и фитосанитарному надзору и (или) действий (бездействия) ее должностных лиц жалоба рассматривается руководителем Федеральной службы по ветеринарному и фитосанитарному надзору.
Жалоба на решение, действия (бездействие) должностных лиц Министерства промышленности и торговли Российской Федерации или Федеральной службы по ветеринарному и фитосанитарному надзору и ее территориальных органов может быть подана в течение 30 календарных дней со дня, когда контролируемое лицо узнало или должно было узнать о нарушении своих прав.
44. Жалоба на предписание Министерства промышленности и торговли Российской Федерации или Федеральной службы по ветеринарному и фитосанитарному надзору Российской Федерации и ее территориальных органов может быть подана в течение 10 рабочих дней со дня получения контролируемым лицом предписания.
45. Жалоба, поступившая в Министерство промышленности и торговли Российской Федерации или Федеральную службу по ветеринарному и фитосанитарному надзору и ее территориальные органы, подлежит регистрации не позднее следующего рабочего дня со дня ее поступления. Жалоба рассматривается в течение 20 рабочих дней со дня ее регистрации.
При рассмотрении жалобы Министерство промышленности и торговли Российской Федерации или Федеральная служба по ветеринарному и фитосанитарному надзору и ее территориальные органы используют информационную систему досудебного обжалования контрольной деятельности.
46. В случаях, установленных частью 1 статьи 42 Федерального закона "О государственном контроле (надзоре) и муниципальном контроле в Российской Федерации", уполномоченное должностное лицо Министерства промышленности и торговли Российской Федерации или уполномоченное лицо Федеральной службы по ветеринарному и фитосанитарному надзору и ее территориальных органов принимают решение об отказе в рассмотрении жалобы в течение 5 рабочих дней с момента получения жалобы.