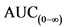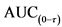СОВЕТ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 3 ноября 2016 г. N 85
ОБ УТВЕРЖДЕНИИ ПРАВИЛ
ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ В РАМКАХ ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА
В соответствии с пунктом 2 статьи 4 и статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, пунктом 86 приложения N 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 98, и Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 108 "О реализации Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза" Совет Евразийской экономической комиссии решил:
1. Утвердить прилагаемые Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза.
2. Настоящее Решение вступает в силу по истечении 10 календарных дней с даты вступления в силу Протокола, подписанного 2 декабря 2015 года, о присоединении Республики Армения к Соглашению о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, но не ранее чем по истечении 10 календарных дней с даты официального опубликования настоящего Решения.
Члены Совета Евразийской экономической комиссии:
|
От Республики Армения
В.ГАБРИЕЛЯН
|
От Республики Беларусь
В.МАТЮШЕВСКИЙ
|
От Республики Казахстан
А.МАМИН
|
От Кыргызской Республики
О.ПАНКРАТОВ
|
От Российской Федерации
И.ШУВАЛОВ
|
Утверждены
Решением Совета
Евразийской экономической комиссии
от 3 ноября 2016 г. N 85
ПРАВИЛА
ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ В РАМКАХ ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА
I. Общие положения
1. Настоящие Правила устанавливают требования к разработке дизайна исследований биоэквивалентности (общего плана исследований, описания способов проведения исследований в зависимости от отбора и формирования групп субъектов исследований, маскирования данных), проведению исследований биоэквивалентности и анализу их результатов, а также основания для замены исследований in vivo исследованиями in vitro.
Цель проведения исследований биоэквивалентности - доказать эквивалентность воспроизведенного (гибридного) лекарственного препарата референтному лекарственному препарату по качеству, чтобы экстраполировать результаты доклинических испытаний и клинических исследований, проведенных в отношении референтного лекарственного препарата, на воспроизведенный (гибридный) лекарственный препарат. Проведение исследований биоэквивалентности требуется при внесении изменений в регистрационное досье зарегистрированного лекарственного препарата (в частности, при изменении состава вспомогательных веществ, технологии производства, места производства, укрупнении или разукрупнении промышленной серии и т.д.), на предрегистрационном этапе при существенном изменении состава, технологии производства лекарственного препарата (если основные доклинические и клинические исследования проведены с неизмененным лекарственным препаратом и необходимо экстраполировать полученные данные о безопасности и эффективности на измененный лекарственный препарат), при изменении лекарственной формы с немедленным высвобождением на лекарственную форму с модифицированным высвобождением, разработке комбинированных лекарственных препаратов и в иных случаях.
2. Два лекарственных препарата, содержащих одинаковое количество действующего вещества, считаются биоэквивалентными, если они являются фармацевтически эквивалентными или фармацевтически альтернативными и их биодоступность (по скорости и степени) после применения в одинаковой молярной дозе укладывается в заранее установленные допустимые пределы. Указанные пределы устанавливаются для обеспечения сопоставимости биофармацевтических свойств лекарственной формы, в которой выпускаются лекарственные препараты in vivo (то есть сопоставимости их по эффективности и безопасности).
3. Для определения скорости и степени абсорбции в исследованиях биоэквивалентности обычно используется кривая "концентрация - время". Следующие фармакокинетические параметры и заранее установленные границы их допустимых отклонений позволяют судить о биоэквивалентности сравниваемых лекарственных препаратов путем оценки их сравнительной биодоступности:
площадь под кривой "концентрация - время" (AUC), отражающая величину экспозиции;
максимальная концентрация вещества (Cmax) в крови, плазме или сыворотке (далее также - плазма, биожидкость);
время достижения максимальной концентрации в биожидкости (tmax).
При этом Cmax и tmax являются параметрами, на которые оказывает влияние скорость абсорбции действующего вещества из лекарственной формы.
4. Настоящие Правила распространяются на лекарственные препараты в виде твердых лекарственных форм для приема внутрь с немедленным высвобождением действующего вещества, содержат требования к планированию и проведению исследований биоэквивалентности путем изучения сравнительной биодоступности разновидностей этих лекарственных форм с немедленным высвобождением, а также других видов лекарственных форм в соответствии с общими требованиями согласно приложению N 1.
Разработка дизайна и проведение исследований, анализ данных сравнительной биодоступности для подтверждения биоэквивалентности лекарственных форм таких лекарственных препаратов проводятся в соответствии с требованиями раздела III настоящих Правил. Если биоэквивалентность невозможно подтвердить с помощью исследований биодоступности, проводятся фармакодинамические или клинические исследования, в соответствии с требованиями согласно приложениям N 2 и 3.
5. В настоящих Правилах устанавливаются критерии, в соответствии с которыми проведение исследований биодоступности in vivo не требуется (для дополнительных дозировок - в соответствии с подразделом 7 раздела III настоящих Правил, для отдельных видов лекарственных форм - в соответствии с приложением N 1, для процедуры биовейвер, основанной на биофармацевтической системе классификации - в соответствии с приложением N 4).
6. При подтверждении биоэквивалентности лекарственных препаратов, которые выпускаются в лекарственных формах с модифицированным высвобождением, трансдермальных лекарственных формах и ингаляционных лекарственных формах, а также лекарственных формах для местного применения и липосомальных лекарственных формах исследования следует проводить в соответствии с актами, входящими в право Евразийского экономического союза (далее - Союз), в сфере обращения лекарственных средств.
7. Сфера применения настоящих Правил ограничена сравнением химических соединений. Порядок сравнения биологических лекарственных препаратов с референтными лекарственными препаратами установлены в правилах проведения исследований биологических лекарственных средств в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией (далее - Комиссия). Подтверждение биоэквивалентности может проводиться в отношении растительных лекарственных препаратов, но основные требования, изложенные в настоящих Правилах, не применимы к растительным лекарственным препаратам, для которых действующие вещества не в полной мере охарактеризованы.
8. Настоящие Правила используются при подаче заявлений о регистрации лекарственных препаратов в рамках Союза.
9. Исследуемые лекарственные препараты, используемые в исследовании биоэквивалентности, должны производиться в соответствии с требованиями правил надлежащей производственной практики Евразийского экономического союза, утверждаемых Комиссией, с представлением соответствующего документального подтверждения в регистрационном досье.
Исследования биоэквивалентности, проведенные за пределами территории Союза, должны соответствовать настоящим Правилам и другим актам, входящим в право Союза, в сфере обращения лекарственных средств.
10. По вопросам, не урегулированным в настоящих Правилах, заявители вправе обращаться в Экспертный комитет по лекарственным средствам при Комиссии (далее - Экспертный комитет при Комиссии) за консультацией.
II. Определения
11. Для целей настоящих Правил используются понятия, которые означают следующее:
"биовейвер" (biowaiver) - процедура оценки биоэквивалентности лекарственного препарата без проведения исследования in vivo;
"биологическая доступность", "биодоступность" (bioavailability) - скорость и степень, с которой действующее вещество или активная часть молекулы действующего вещества абсорбируются из лекарственного препарата и становятся доступными в месте своего действия. Биодоступность определяют как абсолютный или относительный показатель.
Абсолютную биодоступность действующего вещества (активной части молекулы действующего вещества) в определенной лекарственной форме определяют путем сравнения с биодоступностью этого действующего вещества (активной части действующего вещества) при его внутрисосудистом введении, последняя приравнивается к 100 процентам (например, раствор для приема внутрь в сравнении с раствором для внутривенного введения).
Относительную биодоступность действующего вещества (активной части молекулы действующего вещества) в определенной лекарственной форме определяют путем сравнения с биодоступностью другой лекарственной формы, введенной тем же или другим (но не внутривенным) путем (например, таблетки в сравнении с раствором для приема внутрь).
Основой проведения исследований биоэквивалентности и биодоступности большинства лекарственных препаратов является определение относительной биодоступности.
Биодоступность лекарственных препаратов, не предполагающих всасывания в кровоток, допускается оценивать с помощью параметров, способных отразить скорость и степень доступности действующего вещества или активной части молекулы действующего вещества в месте своего действия;
"биологическая эквивалентность", "биоэквивалентность" (bioequivalence) - отсутствие значимых различий по скорости и степени, с которыми действующее вещество или активная часть молекулы действующего вещества фармацевтических эквивалентов или фармацевтических альтернатив становятся доступными в месте своего действия при введении в одинаковой молярной дозе в схожих условиях в исследовании с надлежащим дизайном. При наличии намеренных различий в скорости (например, некоторые лекарственные формы с модифицированным высвобождением), определенные фармацевтические эквиваленты и фармацевтические альтернативы могут быть признаны биоэквивалентными, если отсутствуют значимые различия в степени, с которой действующее вещество или активная часть молекулы действующего вещества из каждого лекарственного препарата становятся доступными в месте своего действия. Правило применимо только в случае различия в скорости, с которой действующее вещество или активная часть молекулы действующего вещества становятся доступными в месте своего действия, запланированы и отражены в общей характеристике лекарственного препарата, незначимы для достижения эффективной концентрации действующего вещества или активной части молекулы действующего вещества в организме при длительном применении и с медицинской точки зрения признаны незначимыми для лекарственного препарата.
Изучение биоэквивалентности может осуществляться как в условиях in vivo (фармакокинетические, фармакодинамические, клинические исследования), так и in vitro (например, исследования теста сравнительной кинетики растворения);
"биофармацевтическая система классификации" (biopharmaceutics classification system, BCS, БСК) - научный подход, позволяющий разделить действующие вещества лекарственных препаратов на основании степени их растворимости в воде и кишечной проницаемости. Вместе с тестом кинетики растворения для лекарственного препарата БСК учитывает 3 основных фактора, влияющих на скорость и степень абсорбции действующих веществ из лекарственных форм с немедленным высвобождением для приема внутрь: растворение, растворимость и кишечную проницаемость;
"воспроизведенный лекарственный препарат", "генерик" - лекарственный препарат, имеющий такой же качественный и количественный состав действующих веществ (активных фармацевтических субстанций) и ту же лекарственную форму, что и референтный лекарственный препарат, и биоэквивалентность которого референтному лекарственному препарату подтверждается соответствующими исследованиями биодоступности.
Различные соли, эфиры, изомеры, смеси изомеров, комплексы или производные действующего вещества признаются одним и тем же действующим веществом, если их безопасность и (или) эффективность значимо не отличаются. Различные лекарственные формы для приема внутрь с немедленным высвобождением признаются в рамках исследований биодоступности одной и той же лекарственной формой (с биофармацевтической точки зрения);
"гибридный лекарственный препарат" - лекарственный препарат, не подпадающий под определение воспроизведенного лекарственного препарата, приведенного в настоящих Правилах, или, при невозможности подтверждения его биоэквивалентности с помощью исследований биодоступности, а также если действующее вещество (действующие вещества), показания к применению, дозировка, лекарственная форма или путь введения такого лекарственного препарата отличаются от таковых референтного лекарственного препарата, что требует представления результатов доклинических и (или) клинических исследований;
"доза лекарственного препарата" (dose) - количество действующего вещества лекарственного препарата на 1 применение (однократное или многократное применение);
"дозировка лекарственного препарата" (strength) - количественно выраженное содержание действующих веществ в единице дозирования, объема или массы в соответствии с лекарственной формой;
"испытание "растворение" для контроля качества" (quality control dissolution test) - предусмотренное Фармакопеей Союза испытание, проводимое с целью рутинного контроля качества серий лекарственного препарата в виде испытания на растворение с одним контрольным временным периодом для отбора проб для лекарственных препаратов с немедленным высвобождением и с 3 и более контрольными временными периодами для лекарственных препаратов с модифицированным высвобождением;
"комбинированный лекарственный препарат" (fixed-dose combination finished pharmaceutical product, FDC-FPP, КЛП) - готовый лекарственный препарат, содержащий 2 и более действующих веществ (активных фармацевтических субстанций);
"лекарственная форма" (dosage form) - состояние лекарственного препарата, соответствующее способам его введения и применения и обеспечивающее достижение необходимого эффекта;
"наполнители" - разновидность вспомогательных веществ, используемых для придания твердым лекарственным формам заданного размера;
"оригинальный лекарственный препарат" (innovator pharmaceutical product) - лекарственный препарат с новым действующим веществом, который был первым зарегистрирован и размещен на мировом фармацевтическом рынке на основании регистрационного досье, содержащего результаты полных доклинических (неклинических) и клинических исследований, подтверждающих его качество, безопасность и эффективность, эквивалентного по содержанию требованиям, установленным частью I приложения N 1 к Правилам регистрации и экспертизы лекарственных средств для медицинского применения;
"референтный лекарственный препарат", "лекарственный препарат сравнения", "компаратор", "контроль" (comparator product) - лекарственный препарат, который используется в качестве эталона в исследованиях сравнительной биодоступности для нормирования исследуемых параметров;
"тест сравнительной кинетики растворения in vitro" (in vitro equivalence dissolution test, ТСКР) - испытание, включающее в себя сравнение профилей растворения исследуемого лекарственного препарата и референтного лекарственного препарата, как правило, в 3 средах - буферных растворах с pH 1,2; 4,5 и 6,8;
"фармацевтическая эквивалентность", "фармацевтические эквиваленты" (pharmaceutical equivalence) - лекарственные препараты в идентичных лекарственных формах, которые содержат одинаковое количество идентичного действующего вещества (активную фармацевтическую субстанцию), то есть одинаковую соль или эфир одной и той же активной части молекулы действующего вещества или в лекарственных формах с модифицированным высвобождением, требующим создания резервуара или избытка, или в таких формах, как предварительно заполненные шприцы (в которых может варьировать остаточный объем), которые доставляют идентичное количество действующего вещества в течение идентичного периода дозирования. Фармацевтические эквиваленты необязательно содержат одинаковые неактивные ингредиенты. Они удовлетворяют идентичным фармакопейным или иным применимым стандартам по подлинности, дозировке, качеству и чистоте, включая активность и в применимых случаях по однородности содержимого, времени распадения и (или) скорости растворения;
"фармацевтически альтернативные лекарственные препараты (pharmaceutical alternatives)" - лекарственные препараты, содержащие одинаковую активную часть молекулы действующего вещества или его предшественник (прекурсор), (необязательно в одинаковом количестве или лекарственной форме) или одинаковую соль или эфир. Каждый такой лекарственный препарат в индивидуальном порядке удовлетворяет идентичным либо своим собственным соответствующим фармакопейным или другим применимым стандартам по подлинности, дозировке, качеству и чистоте (включая активность и в применимых случаях однородность содержимого, время распадения и (или) скорость растворения);
"фиксированная комбинация доз" (fixed-dose combination, FDC, ФКД) - комбинация 2 и более действующих веществ с установленным соотношением дозировок. ФКД используется для обозначения конкретной комбинации действующих веществ вне зависимости от состава или торгового наименования лекарственного препарата. Комбинация действующих веществ может использоваться как совокупность монокомпонентных лекарственных препаратов, применяемых одновременно, так и в виде готового многокомпонентного лекарственного препарата.
В целях настоящих Правил под значимыми понимаются все исследования лекарственного препарата, которые оказывают влияние на информацию о его безопасности, качестве, эффективности и области возможного клинического применения, заявленной в общей характеристике лекарственного препарата.
III. Требования к дизайну, проведению и оценке
исследований биоэквивалентности
12. Объем и дизайн исследований биоэквивалентности необходимо обосновать физико-химическими и фармакокинетическими свойствами действующего вещества и пропорциональностью состава лекарственного препарата. В частности, следует учитывать линейность фармакокинетики, необходимость проведения исследования в зависимости от приема пищи, анализа энантиомеров, а также целесообразность проведения исследований дополнительных дозировок (в соответствии с требованиями подразделов 4 - 7 настоящего раздела).
13. В модуле 2.7.1 регистрационного досье в формате общего технического документа необходимо представить перечень всех значимых исследований (независимо от их результатов), проведенных с лекарственным препаратом, в том числе исследования биоэквивалентности с целью сравнения заявляемого к регистрации лекарственного препарата (то есть имеющего тот же состав и процесс производства) с референтным лекарственным препаратом (в соответствии с требованиями подраздела 2 настоящего раздела).
В отношении всех проведенных исследований в составе модуля 5 регистрационного досье необходимо представить полные отчеты, за исключением пилотных исследований, для которых, если они проводились, достаточно привести краткие синопсисы, которые оформляются в соответствии с приложением N 2 к правилам надлежащей клинической практики Союза, утверждаемым Комиссией. Полный отчет о пилотных исследованиях, в данном случае, необходимо представить по требованию уполномоченных органов государств - членов Союза (далее - государства-члены). В модуль 2.7 необходимо также включить синопсисы отчетов об исследованиях биоэквивалентности и сравнительной биодоступности, проведенных на стадии разработки лекарственного препарата.
1. Дизайн исследования
14. Дизайн исследования необходимо составить таким образом, чтобы влияние лекарственного препарата на его фармакокинетические параметры можно было отличить от влияния других факторов.
15. Стандартный дизайн предполагает следующее.
При сравнении 2 лекарственных препаратов рекомендуется проводить рандомизированное, двухпериодное, перекрестное в 2 последовательностях исследование с приемом однократной дозы. Периоды должны быть разделены отмывочным периодом, достаточным для снижения концентрации действующего вещества ниже порога биоаналитического определения у всех субъектов в начале второго периода исследования. Обычно для этого достаточно 5 периодов полувыведения.
16. Альтернативный дизайн предполагает следующее.
В некоторых случаях, при условии того, что дизайн исследования и статистический анализ научно обоснованы, можно рассматривать альтернативные общепризнанные дизайны: параллельный - для веществ с длительным t1/2; повторный (репликативный, replicate design) - для веществ с высоко вариабельными фармакокинетическими параметрами (в соответствии с требованиями подраздела 11 настоящего раздела).
17. Если вследствие непереносимости прием однократной дозы здоровыми добровольцами недопустим, а исследование однократной дозы у пациентов провести невозможно, допускается проведение исследования у пациентов с многократным приемом лекарственного препарата.
В редких случаях, когда недостаточная чувствительность аналитического метода препятствует точному определению концентрации в биологической жидкости после приема однократной дозы, а равновесная концентрация достаточно высока для получения точных значений, в качестве альтернативы исследованию с приемом однократной дозы допустимо проведение исследования с многократным приемом лекарственного препарата. Однако, принимая во внимание, что исследования с многократным приемом менее чувствительны для определения различий в Cmax, их проведение допустимо только в том случае, если заявитель сможет однозначно доказать, что чувствительность аналитического метода не может быть улучшена и что после приема однократной дозы лекарственного препарата точно измерить концентрацию исходного соединения невозможно, учитывая, что в исследованиях биоэквивалентности допустимо, представив соответствующие обоснования, использовать дозы, превышающие терапевтические (в соответствии с требованиями подраздела 7 настоящего раздела). Проведение исследования с многократным приемом лекарственного препарата вместо однократного в силу недостаточной чувствительности аналитического метода допустимо только в исключительных случаях.
В исследованиях равновесной концентрации отмывочный период после приема предыдущего лекарственного препарата может перекрывать нарастание концентрации во втором периоде (при условии, что продолжительность такого нарастания достаточно длительная и составляет не менее 5 конечных t1/2).
2. Референтный лекарственный препарат и исследуемый
лекарственный препарат
Референтный лекарственный препарат
18. При выборе референтного лекарственного препарата исходят из следующей последовательности:
а) оригинальный лекарственный препарат, качество, безопасность и эффективность которого были установлены при регистрации в Союзе ("зарегистрированный в Союзе оригинальный препарат");
б) оригинальный лекарственный препарат, зарегистрированный в государстве, где уровень требований к регулированию фармацевтического рынка не ниже уровня, установленного в Союзе, при невозможности выполнения подпункта "а" настоящего пункта;
в) воспроизведенный лекарственный препарат, зарегистрированный в каждом из государств-членов и подтвердивший свою биоэквивалентность оригинальному лекарственному препарату (при одобрении Экспертным комитетом при Комиссии) при невозможности выполнения подпунктов "а" и "б" настоящего пункта;
г) лекарственный препарат, имеющий опыт применения на территории одного из государств-членов не менее 25 лет (при одобрении Экспертным комитетом по лекарственным средствам при Евразийской экономической комиссии) при невозможности выполнения подпунктов "а" - "в" настоящего пункта.
19. При исследовании воспроизведенного (гибридного) лекарственного препарата или внесении изменений и дополнений в регистрационное досье лекарственного препарата в части действующих веществ, дозировки, лекарственной формы и пути введения исследуемый лекарственный препарат сравнивают с соответствующей лекарственной формой и дозировкой референтного лекарственного препарата (если не применимы положения абзаца второго настоящего пункта).
Если оригинальный лекарственный препарат на рынке представлен в нескольких лекарственных формах, в качестве референтного лекарственного препарата рекомендуется использовать ту из них, в виде которой он был впервые зарегистрирован и которая использовалась в клинических исследованиях для подтверждения его эффективности и безопасности.
20. Заявитель обязан обосновать выбор референтного лекарственного препарата для исследования биоэквивалентности с учетом результатов количественного определения содержания действующего вещества и данных о его растворении. В серии, подлежащей использованию в качестве исследуемого лекарственного препарата, количественное содержание (установленное с помощью аналитической методики, предложенной для стандартных испытаний качества исследуемого препарата, включенной в спецификацию (нормативный документ по контролю качества)) не должно отличаться более чем на 5 процентов от серии референтного лекарственного препарата (при отсутствии должных обоснований). Заявитель с помощью ТСКР и количественного определения действующего вещества должен обосновать выбор серии референтного лекарственного препарата, планируемой к изучению в исследовании биоэквивалентности. При выборе серии референтного лекарственного препарата для исследования биоэквивалентности рекомендуется изучить несколько серий референтного лекарственного препарата.
Исследуемый лекарственный препарат
21. Исследуемый лекарственный препарат, подлежащий использованию в исследовании биоэквивалентности, не должен отличаться от лекарственного препарата (по составу, технологии производства, производственному оборудованию), который поступит на фармацевтический рынок Союза, что должно быть рассмотрено и обосновано заявителем.
22. Для твердых лекарственных форм для приема внутрь системного действия действуют следующие правила (полные требования к валидации процесса производства содержатся в правилах надлежащей производственной практики Союза и других актах, входящих в право Союза, в сфере обращения лекарственных средств):
а) в отсутствие должных обоснований исследуемый лекарственный препарат должен быть отобран из серии, составляющей, по меньшей мере, 1/10 промышленной серии, или 100 000 единиц лекарственных форм, в зависимости от того, какой из объемов больше;
б) производство использованных серий лекарственного препарата должно обеспечивать высокую степень уверенности в том, что лекарственный препарат и процесс его производства будут воспроизведены в промышленном масштабе.
Объем серии, предназначенной для подтверждения биоэквивалентности, менее 100 000 единиц возможен при условии, что это предлагаемый объем серийного производства, и последующее масштабирование производственных серий не предполагается;
в) описание свойств и составление спецификации на такие критические показатели качества лекарственного препарата, как растворение, следует осуществлять, используя исследованную серию, т.е. серию, изученную в клинических исследованиях, в отношении которой подтверждена биоэквивалентность;
г) образцы лекарственного препарата из дополнительных опытно-промышленных и (или) промышленных серий, предоставленные на регистрацию, необходимо сравнить с образцами из серии, использованной в исследовании биоэквивалентности; они должны иметь сопоставимые профили растворения in vitro в подходящих условиях ТСКР (согласно приложению N 5).
23. В отношении каждой из первых трех промышленных серий до выпуска их на рынок Союза необходимо провести ТСКР с серией, использованной в исследовании биоэквивалентности. В случае истечения ее срока годности в качестве референтной может быть использована предыдущая промышленная серия, профиль растворения которой был сопоставим с профилем растворения серии, использованной в исследовании биоэквивалентности.
Заявитель должен представить результаты ТСКР первых трех промышленных серий по запросу уполномоченного органа государства-члена. При несовпадении профилей растворения заявитель должен представить результаты ТСКР по собственной инициативе без запроса уполномоченного органа и указать конкретные меры, предпринятые для преодоления возникшей ситуации.
Для прочих лекарственных форм с немедленным высвобождением системного действия, необходимо представить аналогичное подтверждение эквивалентности качества промышленных серий по отношению к исследованной серии.
Упаковка сравниваемых лекарственных препаратов
24. Исследуемый лекарственный препарат и референтный лекарственный препарат необходимо упаковать индивидуально для каждого субъекта и периода исследования перед их отправкой в исследовательский центр или в самом исследовательском центре. Упаковку (включая маркировку) следует осуществлять в соответствии с правилами надлежащей производственной практики Союза, утверждаемыми Комиссией.
25. Необходимо предусмотреть возможность точного установления подлинности лекарственных препаратов, применяемых каждым субъектом в каждом периоде исследования. В связи с этим необходимо подробно документировать упаковку, маркировку и введение лекарственных препаратов субъектам. Такая документация должна содержать описание всех мер, принятых для недопущения и обнаружения ошибок дозирования. Рекомендуется использовать этикетки с отрывным корешком.
3. Субъекты исследования
Количество субъектов
26. Количество субъектов, включенных в исследование биоэквивалентности, должно основываться на должном расчете размера выборки. Количество включенных в анализ субъектов исследования биоэквивалентности должно быть не менее 12.
Выбор субъектов
27. Группа субъектов для проведения исследования биоэквивалентности должна быть подобрана таким образом, чтобы можно было обнаружить клинически значимые различия между лекарственными препаратами, обусловленные различиями в их производстве и (или) качестве. С целью снижения вариабельности результатов, не обусловленной различиями между лекарственными препаратами, исследования необходимо проводить среди здоровых добровольцев, за исключением случаев, когда лекарственные препараты несут очевидную угрозу их здоровью, и делают такие исследования неэтичными. В большинстве случаев проведение исследования среди здоровых добровольцев in vivo для установления различий между сравниваемыми лекарственными препаратами считается приемлемым и позволяет экстраполировать результаты исследования на лиц, для которых одобрено применение референтного лекарственного препарата (лица пожилого возраста, дети, пациенты с почечной или печеночной недостаточностью и т.д.).
28. В протоколе исследования необходимо четко прописать критерии включения и невключения. Возраст субъектов должен быть не менее 18 лет с индексом массы тела, по возможности, 18,5 - 30 кг/м2.
29. Соответствие субъектов условиям отбора необходимо подтвердить лабораторными исследованиями, анамнезом и медицинским осмотром. В зависимости от фармакотерапевтической группы и профиля безопасности лекарственного препарата до, во время и по окончании исследования необходимо провести специальные исследования и принять соответствующие меры предосторожности. Пол субъектов не имеет значения, однако необходимо учитывать риск для женщин детородного возраста. Субъекты, по возможности, должны быть некурящими; алкоголизм и наркомания (в том числе в анамнезе) являются критериями для их невключения в исследование. В некоторых случаях из соображений безопасности или в силу фармакокинетических особенностей необходимо предусмотреть фенотипирование и (или) генотипирование субъектов.
30. При параллельном дизайне исследования сравниваемые группы должны быть сопоставимы по всем значимым переменным, которые могут повлиять на фармакокинетику действующего вещества (включая возраст, массу тела, пол, этническую принадлежность, курение, принадлежность к "быстрым" или "медленным" метаболизаторам). Это важное предварительное условие для подтверждения достоверности результатов таких исследований.
31. Если исследуемое действующее вещество вызывает нежелательные реакции и (или) фармакологические эффекты, представляющие неприемлемые риски для здоровых добровольцев, при условии принятия необходимых мер предосторожности и установления соответствующего наблюдения, допускается включение в исследование пациентов.
4. Проведение исследования
Обеспечение стандартности условий проведения исследования
32. В целях сведения к минимуму вариабельности всех вовлеченных факторов, за исключением факторов, обусловленных свойствами сравниваемых лекарственных препаратов, условия проведения исследования необходимо стандартизировать, в связи с чем стандартизации подлежат рацион, прием жидкости и физические нагрузки.
33. Время приема лекарственного препарата необходимо установить заранее. В отсутствие обоснований субъекты не должны принимать пищу как минимум за 8 час до приема лекарственного препарата. Поскольку прием жидкости может повлиять на прохождение принимаемых внутрь лекарственных препаратов через желудок, исследуемый лекарственный препарат и референтный лекарственный препарат необходимо запивать стандартным объемом жидкости (150 - 250 мл). В течение 1 часа до и 2 часов после этого прием жидкости запрещен, в остальном устанавливается свободный питьевой режим. После приема лекарственного препарата прием пищи ограничивают на 4 часа. Рацион и время приема пищи после приема лекарственного препарата необходимо стандартизировать в течение достаточного периода времени (например, 12 часов).
Если исследование должно проводиться после еды, прием лекарственного препарата и пищи осуществляют в соответствии с общей характеристикой лекарственного препарата используемого референтного лекарственного препарата. Если такие сведения в общей характеристике лекарственного препарата референтного лекарственного препарата отсутствуют, то субъекты должны начать прием пищи за 30 минут до приема препарата (продолжительность приема пищи - 30 минут).
34. Поскольку биодоступность активной части молекулы действующего вещества лекарственной формы может зависеть от длительности прохождения через желудочно-кишечный тракт и интенсивности регионарного кровотока, требуется стандартизация положения тела и физической активности субъекта.
35. В течение определенного периода до и во время исследования субъекты должны воздерживаться от приема пищи и напитков, которые могут повлиять на функцию сердечно-сосудистой, пищеварительной системы, печени и (или) почек (например, алкогольные напитки или некоторые соки, например грейпфрутовый). Субъектам не следует принимать какие-либо сопутствующие лекарственные препараты (включая лекарственные препараты растительного происхождения) в течение соответствующего периода до и во время исследования. При этом применение контрацептивов допускается. Если прием сопутствующих лекарственных препаратов неизбежен и они назначены субъекту для купирования нежелательных явлений (например, головной боли), то в сопроводительных документах необходимо отразить сведения о применении (доза и время применения) и возможном влиянии на исход исследования. В исключительных случаях из соображений безопасности или переносимости всем субъектам назначают сопутствующие лекарственные препараты (например, антагонисты опиоидных рецепторов, противорвотные средства). В этом случае необходимо учитывать возможность лекарственного взаимодействия или влияния на биоаналитическую методику, которые могут сказаться на результатах исследования.
36. Лекарственные препараты, которые в соответствии с общей характеристике лекарственного препарата референтного лекарственного препарата должны применяться только в комбинации с другим лекарственным препаратом (например, некоторые ингибиторы протеазы ВИЧ применяют только в комбинации с ритонавиром), разрешается принимать как отдельно, так и в комбинации с рекомендуемым лекарственным препаратом.
37. При изучении биоэквивалентности эндогенных соединений необходимо контролировать факторы, влияющие на их фоновое содержание (например, строгий контроль принимаемой пищи).
Время отбора образцов
38. Для точного описания профиля "концентрация - время" необходимо отобрать достаточное количество образцов. В целях получения точной оценки максимальной экспозиции необходимо предусмотреть частый отбор образцов вблизи предполагаемого tmax. В частности, схема отбора образцов должна быть составлена так, чтобы Cmax не являлась первой точкой на кривой "концентрация - время". Количество отобранных образцов также должно быть достаточным, чтобы обеспечить надежную оценку длительности экспозиции. Это достигается, когда AUC(0-t) перекрывает не менее 80 процентов от
39. В исследованиях с многократным приемом лекарственного препарата для точного определения
40. Если в качестве биологического материала, в котором определяется содержание действующего вещества, выбрана моча, то ее необходимо собирать в течение не менее 3 t1/2. Однако в соответствии с рекомендациями по отбору образцов плазмы, сбор мочи в течение более 72 часов также не требуется. Для определения скорости экскреции интервалы между отбором образцов в фазе абсорбции должны быть, по возможности, как можно короче (с учетом требований подразделов 5 и 6 настоящего раздела).
41. Для эндогенных соединений схема отбора образцов должна позволить описать их фоновое содержание для каждого субъекта в каждом периоде. Как правило, такое определение возможно путем отбора 2 - 3 образцов до приема лекарственного препарата. Иногда, чтобы учесть циркадные колебания фонового содержания эндогенного соединения, требуется регулярно определять его концентрацию в течение 1 - 2 дней до приема лекарственного препарата (с учетом требований подразделов 5 и 6 настоящего раздела).
42. При обычных обстоятельствах биологической жидкостью, отбираемой для измерения концентрации действующих веществ, должна быть кровь. В большинстве случаев измеряется содержание действующего вещества или его метаболитов в сыворотке или плазме. Если отсутствует возможность измерить содержание действующего вещества в плазме, а действующее вещество экскретируется в неизменном виде с мочой и существует пропорциональная взаимосвязь между концентрациями действующего вещества в крови и моче, в качестве биологического материала может использоваться моча. Объем каждого образца следует изучать по возможности незамедлительно после сбора и вносить результаты в отчет. Количество образцов должно быть достаточным, чтобы провести расчет фармакокинетических параметров. Тем не менее, в большинстве случаев следует избегать использования только данных о выделении действующего вещества с мочой, так как это не позволяет рассчитать tmax и максимальную концентрацию вещества в системном кровотоке.
43. Образцы биологической жидкости необходимо обрабатывать и хранить в условиях, при которых ранее не обнаруживалось разложение анализируемых веществ (в большинстве случаев приемлемым является хранение при температуре не выше -20 °C). Данные условия должны быть включены в отчет по валидации (в соответствии с требованиями подраздела 9 настоящего раздела и приложения N 6). Методология сбора образцов оговаривается в протоколе исследования.
Прием лекарственного препарата натощак или после еды
44. Исследования биоэквивалентности, как правило, проводят натощак, поскольку считается, что это соответствует наибольшей чувствительности для выявления различий между сравниваемыми лекарственными препаратами. Если в общей характеристике лекарственного препарата референтного лекарственного препарата его рекомендуется применять натощак или независимо от приема пищи, то исследование биоэквивалентности проводят натощак. Если согласно общей характеристике лекарственного препарата референтного лекарственного препарата его следует применять исключительно после еды, то исследование биоэквивалентности проводят после приема пищи.
Однако для некоторых лекарственных форм (например, микроэмульсии, твердые дисперсии) исследование биоэквивалентности проводят как натощак, так и после приема пищи. Указанное правило не применяется, если лекарственный препарат необходимо принимать либо строго натощак, либо строго после еды.
45. Если требуется проведение обоих видов исследований, то допустимо проводить 2 отдельных перекрестных исследования в 2 группах субъектов или 1 перекрестное исследование в 4 группах субъектов.
46. В условиях, когда прием лекарственного препарата осуществляется после приема пищи, ее состав должен соответствовать рекомендациям общей характеристики лекарственного препарата референтного лекарственного препарата. Если в ней отсутствуют какие-либо рекомендации по этому поводу, то пища должна быть высококалорийной (800 - 1000 ккал), с высоким содержанием жиров (около 50 процентов от общей калорийности). На белки должно приходиться 150 ккал, на углеводы - 250 ккал и на жиры - 500 - 600 ккал. Необходимо описать состав пищи относительно содержания в ней белков, жиров и углеводов в граммах, абсолютном и относительном содержании калорий.
5. Исследуемые параметры
Фармакокинетические свойства
47. При оценке фармакокинетических свойств необходимо использовать фактическое время отбора образцов. В исследованиях биоэквивалентности после однократного приема лекарственного препарата определяют AUC(0-t),
Для лекарственных препаратов с немедленным высвобождением в исследованиях биоэквивалентности в равновесном состоянии необходимо определять
48. При использовании в качестве биологического материала мочи необходимо определять Ae(0-t) и, по возможности, Rmax.
49. Для определения фармакокинетических свойств в исследованиях биоэквивалентности используют внемодельные методы. Использование камерных моделей неприемлемо.
6. Исходное соединение или его метаболиты
Общие принципы
50. В большинстве случаев оценку биоэквивалентности необходимо проводить путем определения концентрации исходного соединения, поскольку для обнаружения различий между лекарственными препаратами по скорости абсорбции Cmax исходного соединения обычно является более чувствительным показателем, чем Cmax его метаболита.
Неактивные пролекарства
51. Для неактивных пролекарств исследование биоэквивалентности следует проводить в отношении исходного соединения. Определять концентрацию активного метаболита не требуется. Однако концентрация в биологических жидкостях некоторых пролекарств достаточно низкая, и они быстро элиминируются из кровотока, что затрудняет подтверждение биоэквивалентности по исходному соединению. В этом случае допускается подтверждать биоэквивалентность для основного активного метаболита без измерения концентрации исходного соединения. В настоящих правилах под исходным соединением, являющимся неактивным пролекарством, понимаются соединения с очень низкой клинической эффективностью или полным ее отсутствием.
Использование данных о метаболите вместо данных об активном
исходном соединении
52. Использование сведений о метаболите вместо данных об активном исходном соединении не рекомендуется. Такая замена допустима лишь в том случае, если заявитель сможет доказать, что чувствительность аналитического метода в отношении исходного соединения не может быть улучшена и что после однократного приема лекарственного препарата точно измерить концентрацию исходного соединения невозможно, учитывая то, что в исследованиях биоэквивалентности допустимо использовать дозы, превышающие максимальные разовые дозы (с учетом требований подраздела 7 настоящего раздела). Замена данных об исходном соединении данными о его метаболите допустима лишь в исключительных случаях. При осуществлении такой замены, заявитель обязан представить все имеющиеся сведения, подтверждающие, что экспозиция метаболита (выраженная в виде AUC) отражает экспозицию исходного соединения и что в терапевтических дозах образование метаболита не является насыщаемым процессом.
Энантиомеры
53. Как правило, допускается использовать нестереоспецифичные биоаналитические методы. Однако при выполнении всех нижеперечисленных условий необходимо измерять концентрацию каждого энантиомера:
энантиомеры обладают различными фармакокинетическими свойствами;
фармакодинамические свойства энантиомеров существенно различаются;
отношение экспозиции энантиомеров (выраженной в виде AUC) меняется при изменении абсорбции.
54. Если все указанные условия выполняются или сведения о них отсутствуют, то необходимо измерять концентрацию каждого энантиомера. Если только один из энантиомеров обладает фармакологической активностью (фармакологическая активность второго энантиомера низкая или полностью отсутствует), то достаточно подтвердить биоэквивалентность только для активного энантиомера.
Использование мочи в качестве биологического материала
55. Если достоверно определить профиль "концентрация - время" в плазме исходного соединения невозможно, то для определения величины экспозиции в качестве замены концентрации в плазме допустимо использование данных об экскреции с мочой. Однако необходимо четко обосновать использование данных мочи при определении максимальной экспозиции. Если удается получить достоверные сведения о Cmax в плазме, то для оценки биоэквивалентности эти данные необходимо представить наряду с величиной экспозиции, полученной при использовании мочи. При использовании мочи в качестве биологического материала заявитель обязан представить все имеющиеся сведения, подтверждающие, что экскреция с мочой отражает экспозицию в плазме.
Эндогенные вещества
56. Если исследуемое вещество является эндогенным, то измерение фармакокинетических параметров необходимо осуществлять с поправкой на его фоновое содержание, чтобы исследуемые фармакокинетические параметры относились к дополнительным концентрациям, полученным вследствие приема лекарственного препарата. При условии приемлемой переносимости и если концентрацию, превосходящую фоновую и достигаемую после приема лекарственного препарата, можно достоверно измерить, в исследованиях биоэквивалентности эндогенных веществ допустимо применение доз, превышающих максимальные разовые дозы. Если после приема различных доз эндогенного вещества разница в экспозиции ранее не была показана, ее необходимо определить либо в пилотном исследовании, либо в рамках одного из периодов основного исследования биоэквивалентности с использованием различных доз референтного лекарственного препарата при условии, что использование этих доз позволит определить потенциальные различия между лекарственными препаратами.
В протоколе исследования необходимо заранее определить и описать метод, используемый для поправки на фоновое содержание эндогенного вещества. В качестве поправки предпочтительно использовать стандартное вычитание: вычитается либо средняя концентрация эндогенного вещества, определенная до приема препарата, либо средняя AUC. Изредка, когда концентрация эндогенного вещества после приема лекарственного препарата существенно превышает фоновую, поправка на фоновое содержание эндогенного вещества не требуется.
57. В исследованиях биоэквивалентности эндогенных веществ напрямую оценить влияние эффекта переноса не представляется возможным, поэтому необходимо соблюдать особую осторожность при выборе длительности отмывочного периода.
7. Исследуемые дозировки
58. Если регистрации подлежат несколько дозировок, то в зависимости от пропорциональности состава между различными дозировками и другими свойствами лекарственного препарата, исследование биоэквивалентности достаточно провести в отношении одной или двух дозировок. Выбор дозировки (дозировок) зависит от линейности фармакокинетики действующего вещества.
59. Если фармакокинетика нелинейна (увеличение AUC непропорционально принимаемой дозе), пригодность различных дозировок для определения потенциальных различий между сравниваемыми лекарственными препаратами может отличаться. Линейность фармакокинетики признается в том случае, если разница между скорректированными по дозе средними AUC для исследуемой дозировки (дозировки, использованной в исследовании биоэквивалентности) и дозировки (дозировок), в отношении которой(ых) проведение исследования биоэквивалентности не планируется, не превышает 25 процентов. Для оценки линейности заявитель должен изучить и критически оценить всю доступную научную литературу на предмет пропорциональности дозы. Линейность констатируется, если различия между скорректированными по дозе AUC находятся в пределах
Если биоэквивалентность для дозировок, обладающей наибольшей чувствительностью в отношении установления различий между сравниваемыми лекарственными препаратами, подтверждена, то в проведении исследований биоэквивалентности in vivo с другими дозировками нет необходимости.
Общие критерии биовейвера для различных дозировок
лекарственного препарата
60. В случае если заявление об отсутствии необходимости проведения исследования биоэквивалентности в отношении дополнительных дозировок (биовейвер), должны соблюдаться следующие условия:
а) производственный процесс лекарственных препаратов с различными дозировками является одинаковым;
б) качественный состав лекарственного препарата с различными дозировками совпадает (данное требование не касается красителей и ароматизаторов);
в) состав лекарственных препаратов с различными дозировками должен быть количественно пропорционален: отношения между содержанием действующего вещества (действующих веществ) и каждого из вспомогательных веществ совпадает для всех дозировок (данное требование не касается оболочек лекарственных препаратов с немедленным высвобождением, оболочек капсул, красителей и ароматизаторов). Если количественная пропорциональность состава отсутствует, то указанное условие считается выполненным, если в отношении исследуемой дозировки и дозировок, для которых не предполагается проведение исследования биоэквивалентности, соблюдаются условия "i" и "ii" или "i" и "iii":
i) содержание действующего вещества (действующих веществ) не превышает 5 процентов от массы ядра таблетки, массы содержимого капсулы;
ii) содержание вспомогательных веществ ядра таблетки или содержимого капсулы совпадает для всех регистрируемых дозировок, изменяется лишь содержание действующего вещества;
iii) содержание наполнителей изменяется в зависимости от содержания действующего вещества; содержание остальных вспомогательных веществ ядра или содержимого капсулы для рассматриваемых дозировок остается неизменным;
г) данные о ТСКР подтверждают отсутствие необходимости в проведении дополнительного исследования биоэквивалентности in vivo.
Линейная фармакокинетика
61. Если описанные в пункте 60 настоящих Правил условия выполняются, достаточно проведения исследования биоэквивалентности в отношении одной дозировки.
62. Как правило, исследование биоэквивалентности проводится для наибольшей дозировки. Для лекарственных препаратов с линейной фармакокинетикой при условии высокой растворимости действующего вещества исследование биоэквивалентности допустимо проводить с использованием меньших дозировок. Выбор меньшей дозировки также может быть обоснован с позиций безопасности или переносимости, когда применение наибольшей дозировки у здоровых добровольцев неприемлемо. Кроме того, если чувствительность аналитического метода не позволяет точно измерить концентрацию при приеме наибольшей дозировки, допускается применение более высокой дозы (предпочтительно использовать несколько таблеток с наибольшей дозировкой). Превышение максимальной терапевтической дозы допускается лишь в том случае, если она хорошо переносится здоровыми добровольцами и отсутствуют ограничения по степени абсорбции или растворимости лекарственного препарата, принятого в такой дозе.
Нелинейная фармакокинетика
63. Если в терапевтическом диапазоне степень увеличения AUC лекарственных препаратов с нелинейной фармакокинетикой больше степени увеличения дозы, исследование биоэквивалентности обычно проводится с использованием наибольшей дозировки. Как и в случае с лекарственными препаратами с линейной фармакокинетикой, выбор меньшей дозировки может быть обоснован с позиций безопасности и переносимости, когда применение наибольшей дозировки у здоровых добровольцев неприемлемо. Вследствие низкой чувствительности аналитического метода аналогично лекарственным препаратам с линейной фармакокинетикой также допускается применение более высоких доз лекарственных препаратов с нелинейной фармакокинетикой.
64. Исследование биоэквивалентности лекарственных препаратов, у которых AUC в терапевтическом диапазоне увеличивается меньше, чем соответствующее увеличение дозы, в большинстве случаев требуется проводить для наибольшей и наименьшей дозировок (или для дозировки, фармакокинетика которой находится в линейном диапазоне), то есть в этом случае проводится 2 исследования биоэквивалентности. Если нелинейность не обусловлена низкой растворимостью, а объясняется, например, насыщением переносчиков и соблюдаются условия, указанные в пункте 60 настоящих Правил) и сравниваемые лекарственные препараты не содержат вспомогательных веществ, влияющих на моторику желудочно-кишечного тракта или белки-переносчики, достаточно проведения исследования биоэквивалентности с наименьшей дозировкой (или дозировкой, фармакокинетика которой находится в линейном диапазоне). Выбор других дозировок может быть обоснован низкой чувствительностью аналитического метода, когда проведение исследования с наименьшей дозировкой невозможно или применение наибольшей дозировки у здоровых добровольцев неприемлемо с позиций безопасности или переносимости.
Исследование крайних вариантов (брекетинг)
65. Если исследование биоэквивалентности требуется провести более чем для 2 дозировок, например, вследствие различий в пропорциональности состава, используют подход, позволяющий ограничиться проведением исследований крайних вариантов. Если выбранные дозировки представляют собой крайние значения, например, максимальная и минимальная дозировки или дозировки, наиболее резко отличающиеся по составу (то есть отличия по составу других дозировок укладываются в эту разность), то допустимо проведение 2 исследований биоэквивалентности.
66. Если оценку биоэквивалентности необходимо осуществить натощак и после приема пищи для 2 дозировок вследствие нелинейной абсорбции или отклонений от пропорциональности состава, достаточно провести исследование натощак и после приема пищи 1 дозировки. Отсутствие необходимости проведения исследования натощак или после приема пищи для других дозировок может быть обосновано данными научной литературы и (или) данными о фармакокинетике, полученными при изучении исследуемой дозировки из других исследований, проведенных натощак и после приема пищи. При выборе условий проведения исследований (натощак или после приема пищи) для изучения остальных дозировок предпочтение отдается условиям, обладающим наибольшей чувствительностью в выявлении возможных различий между сравниваемыми лекарственными препаратами.
Комбинированные лекарственные препараты
67. В отношении всех комбинированных лекарственных препаратов должны выполняться условия пропорциональности состава, предусмотренные настоящими Правилами. При расчете содержания каждого действующего вещества комбинации остальные действующие вещества должны рассматриваться в качестве вспомогательных веществ. Каждый слой двухслойных таблеток может рассматриваться независимо.
8. Методология биоаналитической части исследования
68. Биоаналитическая часть исследований биоэквивалентности должна осуществляться в соответствии с приложением N 6 к настоящим Правилам.
Для получения надежных результатов, поддающихся удовлетворительной интерпретации, необходимо подробно описать используемые биоаналитические методики, полностью их валидировать и документировать. В каждом аналитическом цикле в рамках исследования необходимо подтвердить пригодность методики с использованием образцов для контроля качества.
69. Основными характеристиками биоаналитической методики для обеспечения приемлемости и достоверности полученных аналитических данных являются селективность, нижний предел количественного определения, функция отклика (форма градуировочной кривой), правильность, прецизионность и стабильность.
70. Поскольку поддающаяся обнаружению концентрация анализируемого вещества до приема лекарственного препарата должна составлять 5% от Cmax и менее, нижний предел количественного определения методики должен обеспечивать определение концентрации
71. В протоколе исследования необходимо предусмотреть возможность проведения повторного анализа исследуемых образцов до фактического начала такого анализа. В обычных условиях повторный анализ образцов по фармакокинетическим причинам не допустим, что особенно важно для исследований биоэквивалентности, поскольку это может исказить результаты исследования.
Лица, осуществляющие анализ образцов, не должны знать о принимаемых субъектами исследуемых препаратах.
9. Оценка, анализ и представление результатов исследования
72. Поправку на различия в количественном определении между сериями исследуемого и референтного лекарственных препаратов в исследованиях биоэквивалентности для фармакокинетических параметров вводить, как правило, не допускается. Однако в исключительных случаях, если различия между сериями референтного и исследуемого лекарственных препаратов не превышают 5 процентов (с учетом требований подраздела 2 настоящего раздела), такая поправка допустима. Поправку, наряду с результатами количественного определения исследуемого и референтного лекарственного препарата, необходимо отразить в протоколе исследования.
Отбор субъектов для анализа результатов исследования
73. В статистический анализ необходимо, по возможности, включить всех субъектов, принявших лекарственный препарат. Однако субъекты, участвовавшие в перекрестном исследовании, у которых отсутствуют данные как по исследуемому лекарственному препарату, так и по референтному лекарственному препарату, или субъекты, участвовавшие в параллельном исследовании, у которых отсутствуют данные единственного периода, не должны включаться в анализ.
Обработку данных всех субъектов, принимавших лекарственный препарат, необходимо осуществлять одинаковыми методами. В протоколе исследования не допускается предусматривать включение в анализ данных о "дублерах" добровольцев только с целью замены данных исключенных субъектов. Даже если в ходе исследования не было выбываний из исследования, необходимо предусмотреть включение в анализ всех субъектов, принявших препарат. Таким образом, включать дублеров, проходящих процедуры исследования биоэквивалентности отдельно от основной выборки, не допускается.
74. В исследовании с более чем 2 группами сравнения (например, трехпериодное исследование с 2 референтными лекарственными препаратами или четырехпериодное исследование при приеме натощак и после приема пищи) анализ по каждой сравниваемой паре необходимо осуществлять лишь после предварительного исключения данных, не относящихся к сравниваемым группам.
Критерии исключения субъектов из анализа
результатов исследования
75. Для объективной оценки результатов рандомизированных исследований необходимо, чтобы наблюдение и ведение всех субъектов осуществлялось по единым правилам. Эти правила не должны зависеть от принимаемого лекарственного препарата или исхода, поэтому решение об исключении субъекта из статистического анализа необходимо принять до начала лабораторного анализа образцов.
76. Любая причина может являться критерием исключения субъектов, если она заранее описана в протоколе исследования, а решение об исключении принято до начала анализа образцов. Однако вследствие снижения статистической мощности исследования, а также при необходимом минимуме в 12 субъектов следует избегать исключения последних из исследования.
Допустимыми критериями исключения субъектов из исследования являются рвота или диарея, которые способны исказить результаты измерения концентрации анализируемого вещества. В исключительных ситуациях критерием исключения также может служить одновременное применение других лекарственных препаратов.
77. В протоколе исследования необходимо заранее описать критерии исключения субъектов. Если возникает ситуация, трактуемая как критерий исключения, сведения о ней необходимо занести в индивидуальную регистрационную карту в ходе проведения исследования. Исключение субъектов, основанное на заранее предусмотренных критериях, необходимо четко отразить и перечислить в отчете об исследовании.
78. Ввиду невозможности отделить влияние лекарственных препаратов от других факторов, влияющих на фармакокинетику, исключение данных только на основании статистического анализа или по фармакокинетическим причинам не допускается. Исключениями из данного правила являются:
а) субъекты, в плазме которых концентрация референтного лекарственного препарата не определяется или определяется лишь в незначительных количествах. Концентрации анализируемого вещества у субъекта признаются очень низкими, если его AUC не превышает 5 процентов от средней геометрической AUC референтного лекарственного препарата (рассчитанной без учета данных субъекта с выбросами). Исключение данных по этой причине допустимо лишь в единичных случаях, и в целом ставит под сомнение достоверность (валидность) проведенного исследования;
б) субъекты с ненулевой исходной концентрацией анализируемого вещества, превышающей 5 процентов от Cmax. Такие данные необходимо исключить из исследования биоэквивалентности (в соответствии с требованиями подраздела "Эффекты переноса" настоящего подраздела).
79. В отношении лекарственных препаратов с немедленным высвобождением указанные в настоящем подразделе ситуации могут возникать в случае несоблюдения субъектами режима исследования или недостаточном отмывочном периоде. В первом случае необходимо предусмотреть осмотр ротовой полости субъекта, чтобы удостовериться, что лекарственный препарат был проглочен, во втором - предусмотреть достаточный отмывочный период. Биологические образцы субъектов, исключенных из статистического анализа, необходимо проанализировать, а их результаты представить в отчете об исследовании (в соответствии с требованиями подраздела "Представление данных" настоящего подраздела).
80. Согласно разделу 4 "Проведение исследования" настоящих Правил AUC(0-t) должна перекрывать не менее 80 процентов
Анализируемые параметры и допустимые пределы
81. В исследованиях биоэквивалентности с однократным приемом лекарственного препарата к исследуемым фармакокинетическим параметрам относятся: AUC(0-t) или AUC(0-72 ч) соответственно и Сmax. Отношение данных параметров исследуемого лекарственного препарата к референтному лекарственному препарату должно лежать в интервале 80,00 - 125,00 процентов при 90 процентном доверительном интервале. Границы интервалов округляются до двух знаков после запятой.
82. К изучаемым параметрам исследований биоэквивалентности лекарственных препаратов с немедленным высвобождением с определением равновесной концентрации относятся
83. Если в качестве биологического материала используется моча, показатель Ae(0-t) должен лежать в интервале, описанном для AUC(0-t), а Rmax - в интервале для Cmax.
84. Статистическая оценка tmax не требуется. Однако если указывается, что быстрое высвобождение имеет клиническую значимость и влияет на начало действия или приводит к нежелательным реакциям, значимых различий в tmax и его вариабельности между исследуемым и референтным лекарственными препаратами быть не должно.
85. Допустимые пределы биоэквивалентности лекарственных препаратов с узким терапевтическим диапазоном следует сузить (в соответствии с требованиями подраздела 10 настоящего раздела). Для лекарственных препаратов с высокой вариабельностью Cmax в случае наличия соответствующего обоснования эти границы могут быть расширены.
Статистический анализ
86. Главное значение для оценки биоэквивалентности имеет минимизация риска ложноположительного признания биоэквивалентности. Статистический анализ исследования биоэквивалентности должен подтвердить маловероятность клинически значимого различия между биодоступностью исследуемого и референтного лекарственных препаратов. Процедуры статистической обработки следует оговорить в протоколе перед началом сбора данных.
87. В качестве основного критерия биоэквивалентности используют 90 процентные доверительные интервалы для отношения геометрических средних исследуемых фармакокинетических параметров исследуемого лекарственного препарата и референтного лекарственного препарата. Такой подход равносилен 2 односторонним проверкам нулевой гипотезы об отсутствии биоэквивалентности (о бионеэквивалентности) при 5 процентном уровне значимости для каждого теста.
88. Сравнение исследуемых фармакокинетических параметров проводят с помощью дисперсионного анализа (ANOVA). Для этого предварительно проводят логарифмическое преобразование данных (по основанию десятичного или натурального логарифма). После чего проводят дисперсионный анализ и на основе его результатов строят доверительные интервалы (в логарифмической шкале) для поиска различия между сравниваемыми лекарственными препаратами. Полученные доверительные интервалы подвергаются обратному преобразованию, чтобы построить желаемые доверительные интервалы для отношения средних в исходных (не преобразованных) единицах измерения. Использование непараметрических методов статистического анализа не допускается.
89. В протоколе исследования необходимо заранее предусмотреть выбор конкретной статистической модели анализа. Статистический анализ должен принимать во внимание источники вариабельности, способные повлиять на изучаемую переменную. В такой модели дисперсионного анализа принято использовать такие факторы, как последовательность, субъект последовательности, период и лекарственный препарат. В отношении всех этих факторов следует использовать фиксированные, а не случайные эффекты.
90. Общий принцип заключается в построении 90%-ного доверительного интервала для величины
Аналогичную процедуру следует использовать для сравнения параметров, полученных в результате исследования в равновесном состоянии или суммарного выведения с мочой, если это требуется.
91. Следует представить также данные по описательной статистике для показателя tmax. Если tmax считается клинически значимым, среднее значение и диапазон tmax следует сравнить между исследуемым и референтным лекарственным препаратом для исключения клинически значимых различий. Формальное статистическое сравнение требуется редко. Обычно размер выборки не рассчитывается, для получения необходимой статистической мощности для tmax. Если параметр tmax будет подвергаться статистическому анализу, то изучение должно основываться на непараметрических методах и проводиться с использованием непреобразованных данных. Для повышения точности оценки tmax необходимо взять достаточное количество образцов близких к ожидаемым максимальным концентрациям. Для показателей, описывающих фазу элиминации (t1/2), требуется только описательная статистика.
92. Работа с резко выделяющимися значениями (выбросами) проводится в соответствии с требованиями, изложенными в подразделе "Критерии исключения субъектов из анализа результатов исследования" настоящего раздела. Исключение данных только по причинам статистического и фармакокинетического характера не допустимо.
Исследования в нескольких группах
93. Если перекрестное исследование проведено в 2 и более группах субъектов, т.е. разбиение всей выборки на несколько групп, каждая из которых начинает участие в исследовании в разные дни (например, если из логистических соображений единовременно в клиническом центре можно провести исследование с участием ограниченного числа субъектов), в целях отражения многогруппового характера исследования необходимо модифицировать статистическую модель. В частности, в модели необходимо учесть тот факт, что периоды для первой группы отличаются от периодов для второй (и последующих) группы.
94. Если исследование проведено в двух и более группах и эти группы изучались в различных клинических центрах или в одном и том же центре, но были разделены большим промежутком времени (например, месяцами), возникает сомнение относительно возможности объединения результатов, полученных этих группах, в один анализ. Такие ситуации необходимо обсуждать с уполномоченным органом.
Если предполагается проведение исследования в нескольких группах из логистических соображений, об этом необходимо явно указать в протоколе исследования; при этом, если в отчете отсутствуют результаты статистического анализа, учитывающие многогрупповой характер исследования, необходимо представить научное обоснование отсутствия таких результатов.
Эффекты переноса
95. Не допускается использовать результаты проверки на эффект переноса для принятия каких-либо решений, влияющих на анализ (например, анализ данных, полученных только из первого периода исследования). Вероятность переноса может быть напрямую учтена при отборе образца биологической жидкости до приема лекарственного препарата во втором периоде исследования (и, если применимо, в последующих).
96. Если концентрация до приема лекарственного препарата превышает 5% от Cmax, то сведения, полученные от субъекта в данном периоде, исключаются из статистического анализа. Это значит, что в рамках двухпериодного исследования такой субъект выбывает из анализа. Продолжение исследования считается неприемлемым, если число подлежащих анализу субъектов оказалось менее 12. Данный подход не применим к исследованию эндогенных соединений.
Двухэтапный дизайн исследований биоэквивалентности
97. Исследование биоэквивалентности допускается проводить в два этапа. На первом этапе проводится исследование на начальной (первичной) группе субъектов с анализом полученных результатов. Если биоэквивалентность не подтверждается, то можно набрать дополнительную группу и объединить результаты, полученные в обеих группах для окончательного анализа. Если выбран такой подход, то нужно принять определенные меры, чтобы сохранить неизменной вероятность ошибки I рода для всего исследования, при этом статистические критерии остановки исследования необходимо четко определить до его начала. Анализ данных, полученных в ходе первого этапа, можно рассматривать как промежуточный, и оба анализа необходимо проводить по скорректированным уровням значимости. Для доверительных интервалов следует использовать скорректированную вероятность, равную не менее 90%. Например, использование 94,12%-ных доверительных интервалов для обоих анализов на первом этапе и для объединенных данных первого и второго этапов будет приемлемым, однако существует множество других вариантов, и выбор, какой уровень значимости (
98. При анализе объединенных данных, полученных в ходе двух этапов, фактор "этап" необходимо включить в модель дисперсионного анализа.
Представление данных
99. Для каждого из сравниваемых лекарственных препаратов необходимо представить все значения индивидуальных концентраций и фармакокинетических параметров, наряду с данными описательной статистики, включая геометрическое среднее, медиану, арифметическое среднее, стандартное отклонение, коэффициент вариации, максимальные и минимальные значения. Индивидуальные кривые "концентрация-время" следует представить на линейной и логарифмической шкалах. Необходимо описать метод получения фармакокинетических параметров из исходных данных и количество точек в терминальной логарифмической фазе, использованных для оценки константы скорости терминальной элиминации (которая используется для достоверной оценки
100. В качестве основных результатов статистического анализа изученных фармакокинетических параметров следует указывать точечные оценки и 90%-ные доверительные интервалы для отношения средних значений.
Следует также прилагать стандартные результирующие таблицы дисперсионного анализа, включая результаты статистических тестов на все эффекты в использованной модели.
101. Отчет необходимо детализировать настолько, чтобы фармакокинетический и статистический анализы можно было воспроизвести, то есть включить точное время отбора образцов после приема лекарственного препарата, концентрации анализируемых веществ, значения фармакокинетических параметров каждого субъекта в каждом периоде исследования и схему рандомизации.
102. Необходимо подробно описать все случаи выбывания и исключения субъектов из исследования. По возможности, для каждого такого субъекта в отдельном документе необходимо представить данные о концентрации и фармакокинетических параметрах, но не включать их в общий статистический анализ.
Биоаналитическую методику необходимо документировать до начала исследования (предварительная валидация) для последующего формирования валидационного отчета. Необходимо представить биоаналитический отчет в составе итогового отчета исследования биоэквивалентности. Он должен включать краткое описание использованной биоаналитической методики, результаты по всем градуировочным растворам (стандартам) и образцам для контроля качества. Необходимо представить достаточное количество хроматограмм или других исходных данных, охватывающих весь диапазон концентраций для всех градуировочных растворов (стандартов) и образцов для контроля качества, а также испытуемых (активных) образцов (все хроматограммы и другие первичные данные не менее чем от 20% субъектов с соответствующими образцами для контроля качества и градуировочными растворами/стандартами циклов, относящихся к указанным субъектам).
103. Если в отношении определенной дозировки определенного лекарственного препарата проведено несколько исследований, часть из которых подтверждает его биоэквивалентность, а часть нет, всю совокупность данных необходимо рассматривать как единое целое. В расчет необходимо принимать только исследования, указанные в части III настоящих Правил. Наличие исследований, подтверждающих биоэквивалентность, не является поводом не рассматривать исследования, в которых она не подтверждена. Заявитель должен тщательно проанализировать все результаты и обосновать наличие биоэквивалентности. В качестве альтернативы в дополнение к отдельным исследованиям, по возможности, допускается проведение обобщенного анализа всех исследований. Недопустимо обобщать исследования, не подтверждающие наличие биоэквивалентности, если исследования, подтверждающие биоэквивалентность, отсутствуют.
10. Лекарственные препараты с узким
терапевтическим диапазоном
104. Допустимый интервал для AUC лекарственных препаратов с узким терапевтическим диапазоном следует сузить до 90,00 - 111,11%. Поскольку Cmax занимает особое место с точки зрения эффективности, безопасности и мониторинга концентрации анализируемого вещества, допустимый интервал для данного параметра также следует сузить до 90,00 - 111,11%. Привести исчерпывающее определение лекарственных препаратов с узким терапевтическим диапазоном невозможно, поэтому решение об отнесении действующего вещества к этой группе следует принимать, исходя из клинических особенностей действия и применения лекарственного препарата (при необходимости привлекая экспертов уполномоченных органов государств - членов Союза и (или) Экспертный комитет по лекарственным средствам при Евразийской экономической комиссии).
11. Лекарственные препараты с высокой вариабельностью
105. Если внутрииндивидуальная вариабельность фармакокинетического параметра превышает 30%, такие лекарственные препараты признаются высоко вариабельными. Если заявитель считает, что лекарственный препарат может обладать высокой вариабельностью по скорости и (или) степени абсорбции, рекомендуется проводить исследования с повторным (репликативным) перекрестным дизайном.
106. Лекарственные препараты с высокой вариабельностью, для которых большее различие в Cmax считается клинически незначимым (подтвержденное строгим клиническим обоснованием), ее оценка может осуществляться на основании расширенных интервалов. В этом случае критерий приемлемости для Cmax может быть расширен до 69,84 - 143,19%. В целях расширения критерия приемлемости дизайн исследования биоэквивалентности должен быть повторным и в нем необходимо подтвердить, что вариабельность Cmax референтного лекарственного препарата в исследовании действительно превышает 30%. Заявитель должен доказать, что вычисленная внутрииндивидуальная вариабельность достоверна, а не обусловлена выбросами. Возможность расширения допустимого интервала необходимо заранее оговорить в протоколе исследования.
107. Определение степени расширения интервала основано на внутрииндивидуальной вариабельности, полученной по результатам исследования биоэквивалентности с использованием метода биоэквивалентности в среднем с масштабированием (scaled average bioequivalence) согласно формуле:
где:
U - верхняя граница интервала приемлемости;
L - нижняя граница интервала приемлемости;
k - регуляторная константа, принятая за 0,760;
SWR - внутрииндивидуальное стандартное отклонение логарифмически преобразованных значений Cmax лекарственного препарата сравнения.
108. В приведенной таблице представлены границы интервалов признания биоэквивалентности, рассчитанные на основании описанной в пункте 107 настоящих Правил методологии в зависимости от различной степени вариабельности фармакокинетических параметров лекарственного препарата.
|
Внутрииндивидуальный CV (%) <*>
|
Нижняя граница
|
Верхняя граница
|
||||||||||||||||||||||||||||
|
30
|
80,00
|
125,00
|
||||||||||||||||||||||||||||
|
35
|
77,23
|
129,48
|
||||||||||||||||||||||||||||
|
40
|
74,62
|
134,02
|
||||||||||||||||||||||||||||
|
45
|
72,15
|
138,59
|
||||||||||||||||||||||||||||
|
109. Отношение геометрических средних фармакокинетических параметров должно находиться в пределах 80,00 - 125,00%. Расширение приемлемых границ биодоступности на основании внутрииндивидуальной вариабельности не распространяется на AUC, границы которой вне зависимости от вариабельности должны быть ограничены интервалом 80,00 - 125,00%. 110. При повторном дизайне используют 3-х или 4-х периодную перекрестную схему исследования. IV. Тест сравнительной кинетики растворения in vitro 111. Методика выполнения ТКСР и основные требования по использованию фактора подобия (сходимости, f2-критерий) должны соответствовать требованиям, указанным в приложении N 5 к настоящим Правилам. 1. Тест сравнительной кинетики растворения in vitro 112. Необходимо представить результаты ТСКР серий исследуемого лекарственного препарата и референтного лекарственного препарата, использованных в исследовании биоэквивалентности, в трех различных буферных средах (обычно при pH 1,2; 4,5 и 6,8) и среде, подлежащей использованию в выпускающих испытаниях лекарственного препарата (среда для контроля качества, включаемая в спецификацию (нормативный документ по контролю качества лекарственного препарата)). Исследование некоторых лекарственных форм, например, таблеток, диспергирующихся в полости рта, проводят в различных условиях. Отчет о результатах исследования следует представлять в виде профилей доли растворенного количества во времени с указанием средних значений и обобщающих статистик. 113. В отсутствие иных обоснований спецификации (нормативном документе по контролю качества лекарственного препарата) для контроля качества по показателю "Растворение" исследуемого лекарственного препарата следует составлять на основании профиля растворения серии исследуемого лекарственного препарата, подтвердившей биоэквивалентность референтному лекарственному препарату (в соответствии с требованиями приложения N 5 к настоящим Правилам). Если результаты ТСКР, проведенного с различными сериями, не подтверждают ранее доказанную в исследованиях in vivo биоэквивалентность, то опираются на результаты исследований in vivo. Однако необходимо изучить и объяснить причины такого расхождения. 2. Тест сравнительной кинетики растворения в целях 114. Обоснованность непроведения дополнительных исследований биоэквивалентности in vivo необходимо подтвердить выполненным надлежащим образом ТСКР. Если не указано иное, необходимо изучить растворение при различных значениях pH (обычно при pH 1,2; 4,5 и 6,8). Для всех представленных серий необходимо подтвердить сопоставимость профилей растворения in vitro между дополнительными дозировками и дозировками из серии, использованной в исследовании биоэквивалентности, во всех условиях (с учетом требований приложения N 5 к настоящим Правилам). 115. При значениях pH, при которых ни для одной из дозировок не удается достичь полного растворения, условия проведения ТСКР между дозировками могут различаться. Однако для подтверждения того, что это обусловлено свойствами действующего вещества, а не лекарственной формы, необходимо провести сравнение с соответствующей дозировкой референтного лекарственного препарата. Кроме того, заявитель вправе подтвердить сопоставимость профилей для одинаковых доз (например, между двумя таблетками с дозировкой 5 мг и одной таблеткой с дозировкой 10 мг). V. Отчет об исследовании 1. Отчет об исследовании биоэквивалентности 116. Отчет об исследовании биоэквивалентности должен содержать все необходимые сведения о протоколе исследования, проведении исследования и его анализе. Отчет должен быть составлен и подписан исследователем в соответствии с приложением N 1 к правилам надлежащей клинической практики Союза, утверждаемым Комиссией и приложением N 7 к настоящим Правилам. Структура отчета об исследовании биоэквивалентности должна соответствовать приложению N 7 к настоящим Правилам. В отчете необходимо указать фамилии и имена ответственных исследователей, их место работы, место и длительность проведения исследования, сертификаты или заключения, составленные по результатам аудита (при наличии). 117. Отчет должен содержать подтверждение того, что выбор референтного лекарственного препарата соответствует требованиям подраздела 2 раздела III настоящих Правил. В частности, необходимо указать его торговое наименование, дозировку, лекарственную форму, номер серии, производителя, срок годности и страну, в которой был приобретен референтный лекарственный препарат. 118. В отчете необходимо указать наименование, состав, размер и номер серии, дату производства и, по возможности, дату истечения срока годности исследуемого лекарственного препарата. Сертификаты анализа исследуемого и референтного лекарственных препаратов, использованных в исследовании, прикладываются к отчету в виде приложения. 119. Сведения о концентрациях, фармакокинетических параметрах и результатах статистического анализа необходимо представить в объеме, предусмотренном подразделом "Представление данных" подраздела 9 раздела III настоящих Правил. Сокращение фармакокинетических параметров указывается в соответствии с приложением N 8. 2. Прочие требования к представлению 120. Заявитель должен представить подписанный им официальный документ, подтверждающий, что количественный состав и технология производства изученного в исследовании биоэквивалентности лекарственного препарата и лекарственного препарата, поданного на регистрацию, не отличаются. Необходимо приложить сравнительные профили растворения (в соответствии с разделом IV настоящих Правил и приложением N 7). Отчет о валидации биоаналитического метода и аналитический отчет, подготовленный в соответствии с требованиями приложения N 6, необходимо включить в модуль 5 регистрационного досье лекарственного препарата. 121. По запросу необходимо представить данные (например, в виде электронного текстового файла с данными, разделенными запятыми или пробелами, или файла в формате Excel, или в ином формате по согласованию с уполномоченным органом), достаточные для воспроизведения фармакокинетического и статистического анализа, включая данные о времени отбора образцов, концентрации лекарственного препарата, значениях фармакокинетических параметров каждого субъекта в каждом периоде и схеме рандомизации. VI. Объем исследований при внесении изменений 122. При изменении ранее одобренного состава или технологии производства, которые могут повлиять на биодоступность, проводятся исследования биоэквивалентности in vivo, если не представлено иных обоснований. Всякое представленное обоснование должно основываться на общих принципах, в частности, указанных в приложении N 4 к настоящим Правилам, или при установлении приемлемой (уровня A) in vitro - in vivo корреляции (IVIVC). Если биодоступность измененного лекарственного препарата ранее изучена и установлена приемлемая (уровня A) корреляция между фармакокинетическими параметрами in vivo и кинетикой растворения in vitro, при сопоставимости профиля растворения in vitro между измененным лекарственным препаратом и ранее одобренным в тех же условиях испытания, которые использовались для установления корреляции, исследование биоэквивалентности проводить не требуется (в соответствии с приложением N 5). 123. При внесении изменений в регистрационные досье лекарственных препаратов, которые не являются воспроизведенными лекарственными препаратами (например, оригинальные, новые комбинации, хорошо изученное применение), для проведения исследования биоэквивалентности и ТСКР в качестве референтного лекарственного препарата служит ранее одобренный лекарственный препарат с прежним составом, местом производства, упаковкой и т.п. 124. При внесении изменений в досье воспроизведенного или гибридного лекарственного препарата для исследования биоэквивалентности в качестве препарата сравнения (компаратора, контроля) используется имеющаяся на рынке серия референтного лекарственного препарата. Если лекарственный препарат отсутствует на рынке, то сравнение допускается осуществлять с ранее одобренным составом (воспроизведенного или гибридного лекарственного препарата) с представлением соответствующего обоснования. При изменениях, не требующих исследования биоэквивалентности, следует руководствоваться рекомендациями и требованиями актов, входящих в право Союза, в сфере обращения лекарственных средств. Приложение N 1 ОБЩИЕ ТРЕБОВАНИЯ I. Общие положения Если исследуемый лекарственный препарат содержит другую соль, сложный эфир, стереоизомер или их смесь, другое комплексное соединение или производное действующего вещества по сравнению с референтным лекарственным препаратом, то биоэквивалентность необходимо подтвердить с помощью исследований биоэквивалентности in vivo. Однако если действующее вещество исследуемого лекарственного препарата идентично действующему веществу референтного лекарственного препарата (или содержит соли со схожими свойствами, установленными в части III приложения N 4 к правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемым Евразийской экономической комиссией (далее соответственно - правила исследования биоэквивалентности, Союз, Комиссия)), то в некоторых случаях, описанных ниже и в приложении N 4 к Правилам проведения исследований биоэквивалентности, проведение исследований биоэквивалентности in vivo не требуется. II. Лекарственные формы для приема внутрь с немедленным В отсутствие условий для биовейвера (в соответствии с требованиями приложения N 4 к Правилам проведения исследований биоэквивалентности) в отношении таких лекарственных форм, как таблетки, капсулы и суспензии для приема внутрь необходимо проводить исследования биоэквивалентности. В отношении таблеток, диспергирующихся в полости рта, и растворов для приема внутрь применяются специальные, описанные ниже требования. III. Таблетки, диспергирующиеся в полости рта Таблетки, диспергирующиеся в полости рта (далее - ТДП), предназначены для быстрого растворения во рту. Если действующее вещество также растворяется в слюне и способно всасываться через слизистую оболочку ротовой полости, то время приема лекарственного препарата и его контакта со слизистой оболочкой являются важными факторами. После проглатывания высвободившегося действующего вещества из ТДП, покрытых оболочкой, в зависимости от состава лекарственного препарата, всасывание также происходит и в желудочно-кишечном тракте. Если можно подтвердить, что действующее вещество не всасывается из полости рта, а требует проглатывания для абсорбции из желудочно-кишечного тракта, то лекарственный препарат может удовлетворять критериям биовейвера на основании биофармацевтической системы классификации (БКС) (в соответствии с требованиями приложения N 4 к Правилам проведения исследований биоэквивалентности). Если это невозможно подтвердить, то необходимо проводить исследование биоэквивалентности у человека. Если ТДП являются дополнительной (новой) лекарственной формой и (или) расширением линейки дозировок для иного состава лекарственного препарата для приема внутрь, то проводят трехпериодное исследование с целью оценить применение таблеток, диспергирующихся в полости рта, при одновременном приеме с водой или без нее. Однако если биоэквивалентность между ТДП, принятой без воды, и референтным лекарственным препаратом, запитого водой, показана в двухпериодном исследовании, то биоэквивалентность ТДП, запиваемой водой, считается доказанной. Если ТДП по отношению к референтному лекарственному препарату, представляющему собой ТДП, является воспроизведенным или гибридным лекарственным препаратом, при планировании исследования следует придерживаться следующих требований: а) если референтный лекарственный препарат допустимо как запивать, так и не запивать водой, то исследование биоэквивалентности должно проводиться без приема воды, поскольку это больше соответствует способу применения лекарственного препарата в реальных условиях. Это особенно важно, если действующее вещество растворяется и всасывается из полости рта. Если биоэквивалентность без приема воды подтверждена, то биоэквивалентность с одновременным приемом жидкости считается доказанной; б) если референтный лекарственный препарат либо запивают, либо не запивают водой, то исследование биоэквивалентности проводится в соответствующих условиях (со стандартным двухпериодным перекрестным дизайном); в) если референтный лекарственный препарат либо запивают, либо не запивают водой, а исследуемый лекарственный препарат предназначен для обоих способов приема, то сравнение проводят, запивая и не запивая исследуемый лекарственный препарат водой, при этом лекарственный препарат применяется в соответствии с рекомендованным способом (трехпериодное исследование в 3 группах в 6 последовательностях). В исследованиях по изучению ТДП, если последняя не запивается водой, рекомендуется непосредственно перед приемом лекарственного препарата смочить слизистую оболочку полости рта 20 мл воды. Прием жидкости в течение 1 часа после приема лекарственного препарата запрещен. Исследование биоэквивалентности в отношении пленок, диспергирующихся в полости рта, пленок или защечных таблеток, таблеток подъязычных и таблеток жевательных проводится по аналогии с ТДП. Исследование биоэквивалентности необходимо проводить в соответствии с рекомендуемым способом применения исследуемого лекарственного препарата. IV. Растворы для приема внутрь Если исследуемый лекарственный препарат представляет собой водный раствор для приема внутрь и содержит ту же концентрацию действующего вещества, что и зарегистрированный раствор, то проведение исследований биоэквивалентности не требуется. Однако если вспомогательные вещества способны повлиять на моторику желудочно-кишечного тракта (например, сорбитол, маннитол и т.д.), абсорбцию (например, поверхностно активные вещества или соединения, влияющие на белки-переносчики), процесс растворения и всасывания (например, сорастворители) или стабильность действующего вещества in vivo и если различия между содержанием вспомогательных веществ должным образом не обоснованы прочими данными, то проводится исследование биоэквивалентности. Требования к вспомогательным веществам растворов для приема внутрь аналогичны условиям биовейвера (с учетом требований приложения N 4 к Правилам проведения исследований биоэквивалентности). Если биоэквивалентность исследуемого лекарственного препарата, являющегося раствором для приема внутрь, должна быть подтверждена по отношению к другому лекарственному препарату с немедленным высвобождением, то необходимо провести исследование биоэквивалентности. V. Комбинированные лекарственные препараты Требования по проведению исследования представлены в документе Союза по клинической разработке комбинированных лекарственных препаратов. Условия биовейвера в отношении комбинированных лекарственных препаратов изложены в части V приложения N 4 к Правилам проведения исследований биоэквивалентности. VI. Лекарственные формы с немедленным высвобождением Настоящий раздел, в частности, касается ректальных лекарственных форм. В отношении них, как правило, проводятся исследования биоэквивалентности. Если лекарственный препарат представляет собой раствор, который содержит действующее вещество в той же концентрации, что и зарегистрированный лекарственный препарат с тем же качественным и схожим количественным содержанием вспомогательных веществ, возможен биовейвер (при этом могут применяться требования, аналогичные для растворов для приема внутрь). Положения данного подраздела не относятся к лекарственным препаратам для ингаляций, применяемых для лечения бронхиальной астмы и хронические обструктивные болезни легких, а также к гормональным спреям для назального применения. VII. Растворы для парентерального введения Если исследуемый лекарственный препарат является водным раствором для внутривенного введения и содержит то же действующее вещество, что и зарегистрированный лекарственный препарат, то проведение исследования биоэквивалентности, как правило, не требуется. Однако если одно из вспомогательных веществ способно взаимодействовать с действующим веществом (например, с образованием комплексов) или другим образом влиять на его распределение, метаболизм и выведение, требуется проведение исследования биоэквивалентности. Его можно избежать, если сравниваемые лекарственные препараты содержат примерно одинаковое количество вспомогательных веществ и должным образом доказано, что имеющиеся различия в их содержании не влияют на фармакокинетику действующего вещества. При других парентеральных путях введения, например, внутримышечном и подкожном, если исследуемый лекарственный препарат имеет одинаковый тип растворителя (например, водная или масляная среда), содержит то же действующее вещество в той же концентрации и те же вспомогательные вещества в схожих количествах, что и зарегистрированный лекарственный препарат, то проведение исследований биоэквивалентности не требуется. Более того, проведение исследования биоэквивалентности водных растворов с примерно одинаковым содержанием вспомогательных веществ не требуется, если последние не влияют на вязкость. VIII. Липосомальные, мицеллярные и эмульсионные 1. Липосомальные лекарственные формы Фармакокинетические особенности липосомальных препаратов для внутривенного введения требуют особых подходов подтверждения биоэквивалентности в соответствии с правом Союза. 2. Эмульсии Эмульсии, как правило, не подлежат процедуре биовейвера. Однако при соблюдении нижеперечисленных условий процедура биовейвера возможна: а) лекарственная форма не предназначена для контролируемого высвобождения и (или) контролируемого распределения (векторной доставки); б) способ и скорость введения совпадают с таковыми для зарегистрированного лекарственного препарата. В таких случаях качественный и количественный состав лекарственного препарата не должен отличаться от зарегистрированного; необходимо представить обоснованные данные, подтверждающие высокую схожесть физико-химических свойств, включая фракционный состав дисперсной липидной фазы и другие значимые характеристики эмульсии, в том числе поверхностные свойства (например, 3. Лекарственные препараты липидов для внутривенного Если в отношении этих лекарственных препаратов представлены обоснованные данные о сопоставимости физико-химических свойств, возможна процедура биовейвера. Различия в составе могут быть обоснованы свойствами и показаниями к применению таких лекарственных форм. 4. Мицеллообразующие препараты Мицеллярные растворы для внутривенного введения могут рассматриваться как "комплексные" растворы, поэтому они не подпадают под биовейвер. Однако при соблюдении нижеперечисленных условий биовейвер возможен: а) при разведении лекарственного препарата в соответствии с рекомендациями по его способу применения происходит быстрый распад мицелл, а лекарственная форма не предназначена для контролируемого высвобождения или распределения; б) способ и скорость введения совпадают с зарегистрированным лекарственным препаратом; в) вспомогательные вещества не влияют на распределение, метаболизм и выведение действующего вещества. В таких случаях качественный и количественный состав мицеллярного раствора непосредственно перед введением не должен отличаться от зарегистрированного лекарственного препарата; необходимо представить обоснованные данные, подтверждающие схожесть физико-химических свойств. Например, критическая концентрация мицеллообразования, способность лекарственной формы к солюбилизации (например, максимальная добавочная концентрация (Maximum Additive Concentration)), свободная и связанная фракция действующего вещества и размер мицелл. Эти правила также применимы при незначительных изменениях качественного или количественного состава лекарственного препарата, при условии того, что такие изменения не затрагивают качественный или количественный состав поверхностно-активных веществ. IX. Лекарственные формы с модифицированным высвобождением 1. Лекарственные формы с модифицированным высвобождением Лекарственные формы с модифицированным высвобождением для приема внутрь или трансдермального применения требуют проведения исследований биоэквивалентности в соответствии с актами, входящими в право Союза. 2. Лекарственные формы с модифицированным высвобождением При подтверждении биоэквивалентности в отношении суспензий или иных лекарственных форм, предназначенных для модификации высвобождения действующего вещества при его внутримышечном или подкожном введении, применяются требования к подтверждению биоэквивалентности внесосудистых лекарственных форм с модифицированным высвобождением (например, трансдермальных), согласно актам, входящим в право Союза. X. Лекарственные препараты местного действия, применяемые Правила по изучению лекарственных препаратов местного действия (при пероральном, назальном, легочном, глазном, накожном, ректальном, вагинальном и т.д. введении), отражены в других актах, составляющих право Союза. Если исследуемый лекарственный препарат, представляющий собой раствор (например, капли глазные, спрей назальный (за исключением гормональных назальных спреев) или раствор для наружного применения), не отличается по виду среды растворения (водная или масляная) и содержит ту же концентрацию того же действующего вещества, что и зарегистрированный лекарственный препарат, то подтверждать эквивалентность между ними не требуется. Незначительные различия в содержании вспомогательных веществ допустимы, если значимые фармацевтические свойства исследуемого и референтного лекарственного препарата идентичны или аналогичны. Любые качественные или количественные различия в содержании вспомогательных веществ требуют обоснования с позиций их влияния на терапевтическую эквивалентность. При отсутствии оснований способ и пути введения должны соответствовать зарегистрированному лекарственному препарату. Если после местного применения лекарственных препаратов для местного применения в силу системной абсорбции возникает риск системных нежелательных реакций, необходимо измерить системную экспозицию. Необходимо подтвердить, что системная экспозиция исследуемого лекарственного препарата не превышает таковую лекарственного препарата сравнения, т.е. верхняя граница 90% доверительного интервала не должна превышать верхнюю границу приемлемости биоэквивалентности (125,00%). Ни один из лекарственных препаратов местного действия, применяемых местно или наружно, нельзя рассматривать в качестве воспроизведенного лекарственного препарата, поскольку они подпадают под определение гибридного лекарственного препарата. XI. Газы Если лекарственный препарат является газом для ингаляций, исследования биоэквивалентности не требуются. Приложение N 2 ТРЕБОВАНИЯ 1. Фармакодинамические исследования у здоровых добровольцев или пациентов могут быть использованы для установления эквивалентности между двумя лекарственными препаратами в случае, если фармакокинетический подход не применим. Исследование фармакодинамической эквивалентности может понадобиться, когда количественное определение содержания действующего вещества и (или) метаболитов в плазме или моче не может быть проведено с достаточной чувствительностью и прецизионностью. Кроме того, фармакодинамические исследования эквивалентности у человека необходимы, если измерение концентраций действующего вещества не может быть использовано в качестве суррогатных конечных точек для подтверждения эффективности и безопасности конкретного лекарственного препарата, например, для лекарственного препарата, оказывающего местное действие. Вместе с тем исследования местной доступности, основанные на фармакокинетических исследованиях, проведенные отдельно либо совместно с исследованиями растворения in vitro, могут рассматриваться в качестве суррогатных конечных точек для подтверждения эквивалентности с точки зрения биофармацевтического качества и высвобождения в месте действия для некоторых лекарственных препаратов, оказывающих местное действие. Кроме того, исследования биоэквивалентности также необходимы, чтобы подтвердить эквивалентность системной экспозиции (AUC) лекарственных препаратов при изучении системной безопасности. 2. Фармакодинимические исследования не рекомендованы в отношении лекарственных препаратов, действующее вещество которых проникает в системный кровоток. При этом для оценки системной экспозиции и установления биоэквивалентности можно использовать фармакокинетический подход. Это обусловлено тем, что фармакодинамические и клинические конечные точки характеризуются более низкой чувствительностью к выявлению разницы между лекарственными препаратами в отношении их биофармацевтических свойств, высвобождения и абсорбции. Поскольку кривые зависимости "доза - эффект" для фармакодинамических и клинических конечных точек обычно более пологие, чем соответствующие кривые зависимости фармакокинетических параметров от дозы, необходимо доказать достаточную аналитическую чувствительность исследования, то есть способность различать реакцию, полученную при применении разных доз. Важно проводить сравнения в дозах, вызывающих наиболее значимую реакцию, для определения которых может потребоваться проведение пилотного исследования. Фармакодинамические показатели имеют всегда большую вариабельность, чем фармакокинетические данные. Фармакодинамические показатели часто подвержены значительному влиянию эффекта плацебо, который добавляется к вариабельности и сложному дизайну исследования. В результате может потребоваться набор большого числа пациентов для достижения достаточной статистической мощности. В сравнительные фармакодинамические исследования следует включать группу плацебо (третья группа). 3. При планировании, проведении и оценке результатов исследования должны соблюдаться следующие требования: а) измеряемая реакция должна представлять собой фармакологический эффект, характеризующий эффективность и (или) безопасность лекарственного препарата; б) методика оценки фармакологического эффекта должна быть валидирована с точки зрения правильности, прецизионности, воспроизводимости и специфичности; в) исследуемый лекарственный препарат и референтный лекарственный препарат не должны вызывать максимальную реакцию в ходе исследования, поскольку может оказаться невозможным выявить различия между действующими веществами, применяемыми в дозах, которые вызывают эффекты, близкие к максимальным или максимальные, при этом изучение зависимости "доза - эффект" может быть необходимой частью такого исследования; г) реакция должна измеряться количественно, предпочтительно двойным слепым методом и результаты должны регистрироваться с помощью подходящего оборудования, позволяющего воспроизводить и регистрировать результаты повторяющихся измерений, чтобы обеспечить фиксацию (документирование) фармакодинамических эффектов, которые заменяют измерения концентрации в плазме. Если такие измерения невозможны, можно провести регистрацию по валидированным оценочным шкалам. Если данные ограничены качественными показателями (категориальными данными), требуется выполнение специального статистического анализа; д) субъекты, не реагирующие на лекарственный препарат, должны быть исключены из исследования после предварительного скрининга, и в протоколе должны быть указаны критерии, по которым идентифицируются реагирующие и нереагирующие субъекты; е) в дизайне исследования должны быть учтены лежащая в основе патология и анамнез болезни и приведена информация о воспроизводимости исходных условий; ж) следует использовать перекрестный дизайн, однако, если он не пригоден, можно использовать параллельный дизайн. Отмывочный период между периодами применения препарата должен составлять не менее 5 периодов полуисчезновения острого фармакологического эффекта. Продолжительность измерения острого фармакологического эффекта должна составлять не менее 3 периодов полуисчезновения острого фармакологического эффекта. 4. Для воспроизведенного лекарственного препарата и референтного лекарственного препарата принципы формирования выборки должны оставаться такими же, как описано в разделе 3 части III Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией. 5. В исследованиях, в которых регистрируют непрерывные переменные, изменение интенсивности действия лекарственного препарата, наблюдаемое в течение некоторого промежутка времени, может быть описано таким же образом, что и в исследовании для измерения концентрации действующего вещества в плазме. Можно вывести параметры, описывающие площадь под кривой "эффект - время" (AUEC), максимальную реакцию и время, когда происходит эта реакция. 6. Сравнение воспроизведенного лекарственного препарата и референтного препарата может быть выполнено путем: проведения анализа дозовой зависимости или относительной активности, которая определяется как отношение активности исследуемого лекарственного препарата к активности референтного лекарственного препарата; проведения анализа зависимости эффекта, который состоит из подтверждения эквивалентности (как минимум для 2 уровней доз) по фармакодинамической конечной точке. Минимальным требованием для любого из указанных способов является оценка чувствительности. Для оценки чувствительности необходимо изучить не менее двух ненулевых уровней доз, при этом необходимо продемонстрировать, что один уровень превосходит другой. В связи с этим при отсутствии должного обоснования необходимо изучить более одной дозы обоих лекарственных препаратов. Важно, чтобы были изучены дозы, расположенные в верхней части кривой "доза - эффект". Дозы, находящиеся на нижних участках кривой, могут быть неубедительными для подтверждения эквивалентности, так как могут являться субтерапевтическими. В равной степени доза, расположенная на вершине кривой, может оказывать равнозначный эффект по сравнению с более высокими дозами, что также не может служить подтверждением эквивалентности. Необходимо представить результаты с использованием обоих подходов. В обоих случаях для подтверждения эквивалентности полученные доверительные интервалы фармакодинамических параметров исследуемого и референтного лекарственного препарата должны быть расположены в пределах выбранных границ эквивалентности. Для относительной активности необходимо рассчитать 90% доверительные интервалы (как и в исследованиях биоэквивалентности), в то время как 95% доверительные интервалы рассчитываются для анализа зависимости эффекта. Допустимый диапазон, который применяется в фармакокинетических исследованиях, в данном случае может быть неприменим. Для обоих подходов выбранный диапазон эквивалентности должен быть предусмотрен и тщательно обоснован в протоколе исследования. 7. Отчет о проведении фармакодинамического исследования эквивалентности составляется в соответствии с требованиями приложения N 7 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией с учетом особенностей фармакодинамических исследований. Приложение N 3 ТРЕБОВАНИЯ 1. При некоторых обстоятельствах (например, для липосомальных лекарственных препаратов) кривые зависимости "концентрация в плазме - время" непригодны для оценки эквивалентности 2 лекарственных препаратов. Несмотря на то, что фармакодинамические исследования могут быть подходящим инструментом для установления эквивалентности, иногда указанные исследования не могут быть использованы из-за отсутствия значимых фармакодинамических параметров, которые могут быть достоверно измерены. В этом случае для подтверждения эквивалентности 2 лекарственных препаратов необходимо проведение клинических исследований. Предпочтительнее оценивать эквивалентность, проводя фармакокинетические исследования вместо клинических, так как последние обладают меньшей чувствительностью и могут потребовать включения значительного количества субъектов для достижения достаточной статистической мощности (например, для достижения достаточной статистической мощности, позволяющей обнаружить повышение реакции на исследуемый лекарственный препарат по сравнению с плацебо на 20%, требуется 8600 пациентов, а чтобы продемонстрировать снижение риска на 16%, необходимо привлечь 2600 пациентов с инфарктом миокарда). 2. Сравнительные клинические исследования, описанные в настоящих Требованиях, проводятся по 2 основным дизайнам: дизайн клинической эквивалентности и дизайн не меньшей эффективности. 3. Если целью клинического исследования является подтверждение клинической эквивалентности, должны применяются те же статистические принципы, что и для исследования биоэквивалентности. При этом для фармакодинамических и клинических конечных точек вместо обычно применяемых в фармакокинетических исследованиях 90% доверительных интервалов необходимо использовать 95% доверительные интервалы. Число пациентов, включенных в клиническое исследование, будет зависеть от вариабельности измеряемых параметров и допустимого диапазона их колебаний и обычно намного больше, чем это требуется при исследовании биоэквивалентности. В протоколе исследований эквивалентности должны быть четко определены следующие положения: в качестве контрольных параметров выбирают значимые клинические конечные точки, на основании которых могут быть рассчитаны начало проявления реакции организма (если это подлежит измерению и клинически значимо) и ее интенсивность; размеры границ признания эквивалентности следует определять на основе анализа ситуации, принимая во внимание конкретные клинические условия, например, естественное течение заболевания, эффективность доступных методов лечения и выбранные искомые параметры. В отличие от исследования биоэквивалентности (где используется стандартный допустимый диапазон) допустимый диапазон в клинических испытаниях не может быть стандартным для всех групп лекарственных препаратов и определяется для каждого терапевтического класса (и показания к применению) в индивидуальном порядке; в настоящее время общепринятым для указанных клинических исследований эквивалентности является использование статистических методов, основанных на определении доверительных интервалов. Основная задача заключается в том, чтобы исследуемый лекарственный препарат не отличался от референтного более чем на строго заданную величину. 4. В дизайне таких исследований (по возможности) следует предусмотреть применение плацебо. В некоторых случаях целесообразно включить в финальный сравнительный анализ конечные точки по оценке безопасности. 5. Требования к воспроизведенному лекарственному препарату и референтному лекарственному препарату должны соответствовать указанным в подразделе 2 раздела III Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией. Приложение N 4 ТРЕБОВАНИЯ I. Общие положения 1. Биовейвер, основанный на биофармацевтической системе классификации (БКС), направлен на уменьшение количества исследований биоэквивалентности in vivo, то есть он может служить заменой биоэквивалентности in vivo. Проведения исследований биоэквивалентности in vivo можно избежать, если эквивалентность in vivo подтверждается обоснованными данными, полученными in vitro. Биовейвер, основанный на БКС, ограничен лекарственными препаратами для приема внутрь в твердых лекарственных формах системного действия с немедленным высвобождением, содержащими высоко растворимые действующие вещества с предсказуемой абсорбцией у человека, при условии, что эти действующие вещества имеют широкий терапевтический диапазон (с учетом требований подраздела 11 раздела III Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией). При этом он неприменим к подъязычным, защечным лекарственным формам и лекарственным формам с модифицированным высвобождением. В отношении лекарственных форм, диспергирующихся в полости рта, данный подход применим, если исключена абсорбция из полости рта. 2. Биовейвер, основанный на БКС, предназначен для установления биоэквивалентности между определенными исследуемыми и референтными лекарственными препаратами. Принципы концепции биовейвера могут применяться для подтверждения биоэквивалентности воспроизведенных лекарственных препаратов, расширений оригинальных лекарственных препаратов, при внесении изменений в досье, требующих установления биоэквивалентности, для установления биоэквивалентности между лекарственными препаратами, применявшимися в начальных фазах клинических исследований, а также лекарственными препаратами, выводимыми на рынок. II. Общие требования 3. Биовейвер, основанный на БКС, применим к лекарственному препарату с немедленным высвобождением при условии выполнения всех следующих требований: а) действующее вещество хорошо растворимо и подвергается полной абсорбции (I класс по БКС) с учетом требований раздела III настоящих Требований; б) с учетом специальных требований (в соответствии с подразделом 1 раздела IV настоящих Требований) характеристики растворения in vitro исследуемого и референтного лекарственных препаратов определяются как очень быстрые (>85% в течение 15 минут) или быстрые (85% в течение 30 минут); в) качественный и количественный состав вспомогательных веществ, способных повлиять на биоэквивалентность, одинаковый. При этом, целесообразно использовать одинаковые вспомогательные вещества в сопоставимых количествах (в соответствии с требованиями подраздела 3 раздела IV настоящих Требований); г) отсутствуют риски, связанные с вероятностью принять ошибочное заключение о возможности использования процедуры биовейвера, с учетом величины терапевтического индекса и клинических показаний к применению для действующего вещества в составе лекарственного препарата. 4. Биовейвер, основанный на БКС, также применим к лекарственному препарату с немедленным высвобождением при условии выполнения всех следующих требований: а) действующее вещество хорошо растворимо и подвергается ограниченной абсорбции (III класс по БКС) с учетом требований раздела III настоящего приложения); б) с учетом специальных требований (в соответствии с требованиями подраздела 1 раздела IV настоящих Требований) характеристики растворения in vitro исследуемого и референтного лекарственных препаратов определяются как очень быстрые (>85% в течение 15 минут); в) качественный и количественный состав вспомогательных веществ, способных повлиять на биоэквивалентность, одинаковый. При этом, целесообразно использовать одинаковые вспомогательные вещества в сопоставимых количествах (в соответствии с требованиями подраздела 3 раздела IV настоящих Требований); г) отсутствуют риски, связанные с вероятностью принять ошибочное заключение о возможности использования процедуры биовейвера, с учетом величины терапевтического индекса и клинических показаний к применению для действующего вещества в составе лекарственного препарата. 5. Следует более критично подходить к оценке выполнения условий (например, место абсорбции, возможность взаимодействия с белками-переносчиками в месте абсорбции, состав вспомогательных веществ и терапевтические риски) в отношении лекарственных препаратов III класса по БКС, чем к препаратам I класса по БКС. Возможность регистрации лекарственного препарата, действующее вещество которого принадлежит III классу по БКС, без проведения исследований биоэквивалентности in vivo необходимо согласовать с Экспертным комитетом по лекарственным средствам при Евразийской экономической комиссии. III. Действующее вещество 6. В целях описания свойств действующего вещества, подпадающего под концепцию биовейвера, достаточно литературных данных о соединениях, изложенных в реферируемых (цитируемых) научных изданиях и документах уполномоченных органов (организаций) в сфере обращения лекарственных средств. 7. Если действующие вещества исследуемого и референтного лекарственных препаратов одинаковые, возможен биовейвер. Биовейвер также возможен, если исследуемый и референтный лекарственные препараты содержат различные соли при условии их принадлежности к I классу по БКС (высокая растворимость и полная абсорбция в соответствии с требованиями подразделов 1 и 2 раздела III настоящих Требований). Если исследуемый лекарственный препарат содержит сложные эфиры, стереоизомеры и их смеси, комплексы или производные действующего вещества референтного лекарственного препарата, биовейвер невозможен, поскольку различия могут привести к различной биодоступности, не выявляемой с помощью экспериментов, используемых в концепции биовейвера, основанного на БКС. Действующее вещество не должно обладать узким терапевтическим диапазоном (в соответствии с требованиями подраздела 11 раздела III Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией). 1. Растворимость 8. Необходимо установить и проанализировать профиль pH-растворимости действующего вещества. Действующее вещество признается хорошо растворимым, если при температуре 37 2. Всасывание (проникающая способность) 9. При заявлении на регистрацию лекарственного препарата как биовейвер, основанный на БКС, рекомендуется подтвердить полную абсорбцию действующего вещества у человека. С этой целью под полным всасыванием понимают абсорбцию 10. Наличие полного всасывания должно быть обосновано исследованиями у человека. В качестве обоснования допускается использовать результаты исследований: абсолютной биодоступности; материального баланса. При использовании метода материального баланса для вычисления всосавшейся фракции необходимо удостовериться, что метаболиты, учтенные при расчете всосавшейся фракции, образовались после абсорбции. В связи с этим при расчете общей радиоактивности, экскретируемой с мочой, необходимо удостовериться, что в желудочном или кишечном соке не произошла частичная деградация или биотрансформация неизмененного действующего вещества. Реакции I фазы (например, окисление) или II фазы (например, конъюгация) метаболизма могут происходить лишь после абсорбции (не в желудочном или кишечном соке). Таким образом, основываясь на данных исследований материального баланса, всасывание признается полным, если общее содержание исходного соединения в моче и его метаболитов (прошедших I и (или) II фазы метаболизма) в моче и кале составляет 11. Кроме того, высоко растворимые действующие вещества с неполным всасыванием (III класс по БКС) также могут подпадать под биовейвер, если выполняются определенные требования к составу лекарственного препарата и профилю растворения in vitro (в соответствии с подразделом 3 раздела IV настоящих Требований). При отнесении соединений к I классу по БКС и отсутствии обоснованных доказательств в пользу их полного всасывания к ним также предъявляются более жесткие требования (например, проведение исследований биоэквивалентности in vivo, иных клинических исследований). 12. Одним из условий биоэквивалентности между водными растворами и твердыми лекарственными формами любого соединения, принимаемого внутрь, является отсутствие существенных различий в профиле абсорбции, обусловленных различиями в лекарственной форме с быстрым высвобождением. Установленная биоэквивалентность между водной и твердой лекарственными формами немедленного высвобождения некоторого соединения, принимаемого внутрь, принимается в качестве подтверждения, поскольку свидетельствует о том, что ограничение абсорбции, обусловленное свойствами лекарственной формы лекарственного препарата (с немедленным высвобождением), является незначительным. Качественные исследования проницаемости in vitro, в том числе с использованием стандартных образцов, также свидетельствуют в пользу результатов, полученных in vivo. IV. Лекарственный препарат 1. Проведение теста сравнительной кинетики растворения 13. При изучении свойств лекарственного препарата необходимо доказать немедленное высвобождение и сопоставимость исследуемых лекарственных препаратов, то есть подтвердить сопоставимую кинетику растворения in vitro между исследуемым и референтным лекарственными препаратами при физиологических значениях pH в условиях эксперимента. Однако это не позволяет установить корреляцию in vitro/in vivo. Кинетику растворения in vitro следует изучить в диапазоне pH 1,0 - 6,8 (не менее чем при 3 значениях pH: 1,2, 4,5 и 6,8). Дополнительные исследования могут потребоваться при pH с наименьшей растворимостью действующего вещества (следует представить обоснование отсутствия необходимости таких исследований). Использование каких-либо поверхностно-активных веществ не допускается. 14. Исследуемый и референтный лекарственные препараты должны соответствовать требованиям, изложенным в подразделе 2 раздела III Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией. В соответствии с этими требованиями рекомендуется проводить исследование в отношении более чем 1 серии исследуемых лекарственных препаратов. 15. Сравнительные испытания растворения in vitro должны соответствовать требованиям Фармакопеи Евразийского экономического союза. В связи с этим необходимо представить подробное описание условий исследования и аналитических методик, включая данные по их валидации. Для статистической достоверности каждый эксперимент рекомендуется проводить с 12 пробами (образцами) лекарственного препарата. Стандартные условия исследования включают: прибор: лопастная мешалка или корзинка; объем среды растворения: 900 мл или менее; температура среды растворения: 37 скорость вращения: лопастная мешалка - обычно 50 оборотов в минуту, корзинка - обычно 100 оборотов в минуту; схема отбора проб: например, на 10, 15, 20, 30 и 45 минутах; буферные растворы: pH 1,0 - 1,2 (обычно 0,1 M HCl или имитация желудочного сока без ферментов), 4,5 и 6,8 (или имитация кишечного сока без ферментов), pH должна регулярно контролироваться. Рекомендуется использовать буферные растворы по Фармакопее Евразийского экономического союза; прочие условия: отсутствие поверхностно-активных веществ. Применение ферментов допускается в отношении желатиновых капсул или таблеток, покрытых желатиновой оболочкой. 16. Необходимо представить полный аналитический отчет о проведении теста сравнительной кинетики растворения (ТСКР) in vitro, включая протокол исследования, сведения об исследуемых сериях и сериях сравнения, подробное описание экспериментальных условий, результаты валидации использованных методов, индивидуальные и средние значения, а также соответствующие обобщающие статистики и др. в соответствии с приложением N 7 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией. 2. Оценка результатов теста сравнительной кинетики 17. Лекарственные препараты признаются очень быстро растворимыми, если 85% заявленного содержания действующего вещества растворяется в течение 15 минут. В этом случае профили растворения исследуемого и референтного лекарственных препаратов признаются сопоставимыми без дальнейших математических расчетов. Если процесс растворения со степенью высвобождения 85% от заявленного содержания действующего вещества длится более 15 минут, но не превышает 30 минут, то необходимо доказать отсутствие значимых различий (сопоставимость). В целях подтверждения сопоставимости профилей растворения исследуемого и референтного лекарственных препаратов используют критерий f2 (в соответствии с требованиями, установленными приложением N 5 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией) или другие подходящие тесты. При этом объяснение различий в профилях растворения с клинических или терапевтических позиций нецелесообразно, поскольку испытание растворения не отражает корреляцию in vitro/in vivo. 3. Вспомогательные вещества 18. Несмотря на то, что влияние вспомогательных веществ, содержащихся в лекарственных формах с немедленным высвобождением, на биодоступность хорошо растворимых и полностью всасывающихся действующих веществ (то есть относящихся к I классу по БКС) считается маловероятным, его нельзя полностью исключать. В связи с этим во всех случаях (в том числе с действующим веществом I класса по БКС) в исследуемом лекарственном препарате рекомендуется использовать схожие количества тех же вспомогательных веществ, что и в референтном лекарственном препарате. 19. В целях исключения различного влияния на мембранные переносчики одним из условий биовейвера в отношении действующего вещества III класса по БКС является отсутствие различий по качественному и высокая сопоставимость по количественному составу вспомогательных веществ в соответствии с критериями, приведенными в таблице. Рекомендуемые критерии для установления
|