СОВЕТ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 12 февраля 2016 г. N 46
О ПРАВИЛАХ
РЕГИСТРАЦИИ И ЭКСПЕРТИЗЫ БЕЗОПАСНОСТИ, КАЧЕСТВА
И ЭФФЕКТИВНОСТИ МЕДИЦИНСКИХ ИЗДЕЛИЙ
В соответствии с пунктом 2 статьи 31 Договора о Евразийском экономическом союзе от 29 мая 2014 года, пунктами 2 и 4 статьи 4 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года, пунктом 92 приложения N 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 98, и в целях исполнения Решения Высшего Евразийского экономического совета от 23 декабря 2014 г. N 109 "О реализации Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза" Совет Евразийской экономической комиссии решил:
1. Утвердить прилагаемые Правила регистрации и экспертизы безопасности, качества и эффективности медицинских изделий (далее - Правила).
2. Установить, что:
а) в переходный период до 31 декабря 2021 г.:
регистрация медицинского изделия по выбору производителя медицинского изделия (его уполномоченного представителя) может осуществляться в соответствии с Правилами либо в соответствии с законодательством государства - члена Евразийского экономического союза (далее - государства-члены);
медицинские изделия, зарегистрированные в соответствии с законодательством государства-члена, обращаются на территории этого государства-члена;
б) документы, подтверждающие факт регистрации медицинских изделий и выданные уполномоченным органом государства-члена в области здравоохранения в соответствии с законодательством этого государства-члена, действительны до окончания срока их действия, но не позднее 31 декабря 2021 г.
3. Государствам-членам до 31 декабря 2016 г.:
а) утвердить размер сборов (пошлин) или иных обязательных платежей, предусмотренных Правилами, с учетом сложности процедур и объема выполняемых работ, проводимых в референтном государстве и государствах признания, в том числе при:
регистрации медицинского изделия;
экспертизе безопасности, качества и эффективности медицинского изделия;
внесении изменений в регистрационное досье медицинского изделия;
выдаче дубликатов регистрационных удостоверений;
б) определить органы (организации), ответственные за осуществление регистрации, внесение изменений в регистрационное досье и иные связанные с регистрацией медицинских изделий процедуры, предусмотренные Правилами, и проинформировать об этом Евразийскую экономическую комиссию.
4. Настоящее Решение вступает в силу по истечении 10 календарных дней с даты вступления в силу Протокола, подписанного 2 декабря 2015 года, о присоединении Республики Армения к Соглашению о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года, но не ранее чем по истечении 10 календарных дней с даты официального опубликования настоящего Решения.
Члены Совета Евразийской экономической комиссии:
|
От Республики Армения
В.ГАБРИЕЛЯН
|
ОТ Республики Беларусь
В.МАТЮШЕВСКИЙ
|
От Республики Казахстан
Б.САГИНТАЕВ
|
От Кыргызской Республики
О.ПАНКРАТОВ
|
От Российской Федерации
И.ШУВАЛОВ
|
Утверждены
Решением Совета Евразийской
экономической комиссии
от 12 февраля 2016 г. N 46
ПРАВИЛА
РЕГИСТРАЦИИ И ЭКСПЕРТИЗЫ БЕЗОПАСНОСТИ, КАЧЕСТВА
И ЭФФЕКТИВНОСТИ МЕДИЦИНСКИХ ИЗДЕЛИЙ
I. Общие положения
1. Настоящие Правила разработаны в соответствии с пунктом 2 статьи 31 Договора о Евразийском экономическом союзе от 29 мая 2014 года и пунктом 2 статьи 4 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года и устанавливают порядок проведения регистрации и экспертизы безопасности, качества и эффективности медицинских изделий (далее - регистрация и экспертиза медицинского изделия), внесения изменений в регистрационное досье медицинского изделия, выдачи дубликатов регистрационных удостоверений, а также отказа в регистрации медицинского изделия, приостановления и отмены действия (аннулирования) регистрационного удостоверения медицинского изделия в рамках Евразийского экономического союза (далее - Союз).
Требования настоящих Правил не применяются в отношении медицинских изделий, потребность в которых возникает в чрезвычайных ситуациях или для диагностики новых, природно-очаговых или особо опасных инфекционных заболеваний, обращение которых регулируется законодательством государств-членов.
2. В переходный период по выбору производителя медицинских изделий (его уполномоченного представителя) экспертиза и регистрация медицинских изделий осуществляются в соответствии с законодательством государства - члена Союза (далее - государство-член) или с настоящими Правилами.
3. Понятия, используемые в настоящих Правилах, означают следующее:
"безопасность медицинских изделий" - отсутствие недопустимого риска, связанного с причинением вреда жизни, здоровью человека, окружающей среде;
"валидация" - подтверждение посредством представления объективных свидетельств выполнения требований, предназначенных для конкретного использования или применения;
"валидация программного обеспечения" - процесс подтверждения пригодности программного обеспечения для решения конкретных прикладных задач;
"верификация" - подтверждение на основе представления объективных свидетельств выполнения установленных требований;
"верификация программного обеспечения" - процесс подтверждения соответствия программного обеспечения установленным требованиям (в том числе соответствующему техническому заданию, спецификации, отраслевым стандартам);
"государство признания" - государство-член, уполномоченный орган (экспертная организация) которого осуществляет процедуру согласования экспертного заключения референтного государства;
"единый реестр медицинских изделий, зарегистрированных в рамках Евразийского экономического союза" - электронная база данных медицинских изделий, зарегистрированных и разрешенных к медицинскому применению на территории Союза;
"заявитель" - производитель, являющийся резидентом государства-члена, или его уполномоченный представитель;
"идентификация и маркировка операционной системы" - анализ информации об использованной при разработке программного обеспечения операционной системе с целью оценки возможностей защиты программного обеспечения от несанкционированного доступа;
"качество медицинского изделия" - степень соответствия совокупности свойств и характеристик медицинского изделия целям его предназначенного использования;
"классификация медицинского изделия в зависимости от потенциального риска применения" - отнесение или определение принадлежности медицинского изделия к одному из классов потенциального риска применения в медицинских целях;
"комплектующее к медицинским изделиям" - изделие, не являющееся медицинским изделием или принадлежностью к медицинскому изделию, в том числе блоки, части, элементы изделия, материалы, запасные части, предназначенные производителем медицинского изделия для применения в составе медицинского изделия или совместно с медицинским изделием;
"медицинские изделия для диагностики in vitro" - любые инструменты, аппараты, приборы, оборудование, материалы, реагенты, калибраторы, контрольные материалы и прочие изделия, применяемые в медицинских целях отдельно или в сочетании между собой, а также вместе с принадлежностями, необходимыми для применения указанных изделий по назначению (включая специальное программное обеспечение), и предназначенные производителем для применения при исследованиях in vitro образцов биологических материалов человека для получения информации относительно физиологического или патологического состояния, врожденной патологии, предрасположенности к определенному клиническому состоянию или болезни, совместимости тканей с потенциальным реципиентом, прогнозирования реакций на терапевтические воздействия, выбора терапевтических средств и (или) контроля лечения;
"модификация медицинского изделия" - разновидность медицинского изделия, имеющая общие с основным медицинским изделием конструктивные признаки, разработанные на базе основного изделия с целью его усовершенствования, расширения либо специализации применения в медицинских целях;
"мультицентровое исследование программного обеспечения" - одновременное тестирование программного обеспечения в условиях его планируемого применения на нескольких внешних экспериментальных площадках (вне предприятия - разработчика такого программного обеспечения);
"набор (комплект) медицинских изделий" - совокупность медицинских изделий, имеющих единое назначение и маркировку, с указанием перечня указанных медицинских изделий;
"принадлежность" - изделие, не являющееся медицинским изделием, предназначенное производителем для совместного применения с одним или несколькими медицинскими изделиями для использования в соответствии с их назначением;
"производитель медицинского изделия" - юридическое лицо или физическое лицо, зарегистрированное в качестве индивидуального предпринимателя, ответственные за разработку и изготовление медицинского изделия, делающие его доступным для использования от своего имени независимо от того, разработано и (или) изготовлено медицинское изделие этим лицом или от его имени другим лицом (лицами), и несущие ответственность за безопасность, качество и эффективность медицинского изделия;
"производственная площадка" - территориально обособленный комплекс, предназначенный для выполнения всего процесса производства медицинского изделия или его определенных стадий;
"расходный материал к медицинским изделиям" - изделия и материалы, расходуемые при использовании медицинских изделий, обеспечивающие проведение манипуляций в соответствии с функциональным назначением медицинского изделия;
"регистрационное досье" - комплект документов и материалов установленной структуры, представляемый заявителем при регистрации медицинского изделия, внесении изменений в регистрационное удостоверение, а также копии решений, принятых уполномоченным органом (экспертной организацией) в отношении конкретного медицинского изделия;
"регистрационный номер" - кодовое обозначение, присваиваемое медицинским изделиям при их регистрации, под которым они вносятся в единый реестр медицинских изделий, зарегистрированных в рамках Союза, и сохраняемое неизменным при обращении медицинского изделия в рамках Союза;
"регистрационное удостоверение" - документ единой формы, подтверждающий факт регистрации медицинского изделия;
"регистрация медицинского изделия" - процедура выдачи уполномоченным органом разрешения на медицинское применение медицинского изделия в рамках Союза;
"референтное государство" - выбранное заявителем государство-член, уполномоченный орган которого осуществляет регистрацию медицинского изделия;
"уполномоченный представитель производителя" - юридическое лицо или физическое лицо, зарегистрированное в качестве индивидуального предпринимателя, являющиеся резидентами государства-члена и уполномоченные в соответствии с доверенностью производителя медицинского изделия представлять его интересы и нести ответственность в части обращения медицинского изделия в рамках Союза и исполнения обязательных требований, предъявляемых к медицинским изделиям.
4. Регистрация и экспертиза медицинского изделия являются обязательными условиями его выпуска в обращение в рамках Союза и осуществляются уполномоченным органом референтного государства. При этом предъявляются одинаковые требования в отношении медицинских изделий, произведенных на территории Союза и ввезенных на таможенную территорию Союза из третьих государств.
5. До подачи в уполномоченный орган референтного государства заявления на регистрацию и экспертизу медицинского изделия заявитель осуществляет сбор доказательств безопасности и эффективности медицинского изделия и подготовку соответствующего регистрационного досье.
6. В целях подготовки регистрационного досье заявитель:
а) получает предварительные консультации экспертной организации по вопросам регистрации и экспертизы медицинского изделия (при необходимости);
б) проводит технические испытания, испытания (исследования) с целью оценки биологического действия медицинского изделия, испытания в целях утверждения типа средств измерений (в отношении медицинских изделий, отнесенных к средствам измерений, перечень которых утверждается Комиссией) для подтверждения соответствия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них в выбранных заявителем учреждениях и организациях, имеющих право проводить такие (испытания) исследования в целях регистрации медицинских изделий и включенных в единый реестр уполномоченных организаций Союза (далее - уполномоченные организации);
в) проводит клинические испытания (исследования) в соответствии с правилами проведения клинических испытаний (исследований) медицинских изделий в выбранных заявителем уполномоченных организациях либо включает в регистрационное досье имеющиеся клинические данные.
7. В целях регистрации медицинского изделия проводится экспертиза медицинского изделия экспертной организацией, определенной уполномоченным органом государства-члена (далее - экспертная организация).
8. Производитель медицинского изделия обеспечивает внедрение и поддержание системы менеджмента качества этого изделия в соответствии с утверждаемыми Евразийской экономической комиссией (далее - Комиссия) требованиями к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска применения.
9. При регистрации и экспертизе медицинских изделий уполномоченные органы взаимно признают результаты технических испытаний, исследований (испытаний) с целью оценки биологического действия этих медицинских изделий, клинических испытаний, испытаний в целях утверждения типа средств измерений (в отношении медицинских изделий, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений, перечень которых утверждается Комиссией) при условии, что они выполнены в соответствии с требованиями и правилами, установленными Комиссией.
10. Регистрация медицинского изделия осуществляется референтным государством на основании результатов экспертизы медицинского изделия и согласования экспертного заключения государствами признания.
11. Документом, подтверждающим факт регистрации медицинского изделия, является регистрационное удостоверение, форма и правила заполнения которого определены согласно приложению N 1.
Регистрационное удостоверение выдается бессрочно и действует в рамках Союза.
12. Зарегистрированное медицинское изделие должно соответствовать общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них, утверждаемым Комиссией. Ответственность за соответствие медицинских изделий указанным общим требованиям несет производитель медицинского изделия (его уполномоченный представитель).
13. Расходы на регистрацию и экспертизу медицинского изделия несет заявитель в соответствии с законодательством государства-члена.
14. При одновременной подаче на регистрацию нескольких модификаций медицинского изделия, относящихся к одному виду медицинского изделия в соответствии с применяемой в Союзе номенклатурой медицинских изделий, изготовленных одним производителем, отличающихся друг от друга изменениями комплектации и (или) технических параметров, не влияющими на принцип работы и функциональное назначение, относящихся к одному классу потенциального риска применения, заявитель представляет 1 заявление и 1 регистрационное досье. В случае если представленные модификации будут относиться к разным видам медицинского изделия в соответствии с указанной номенклатурой, каждая модификация регистрируется отдельно с предоставлением отдельного регистрационного досье.
15. В случае если медицинское изделие, зарегистрированное в соответствии с настоящими Правилами, заявляется для регистрации в государствах-членах, не указанных заявителем ранее в качестве государств признания, а также в государствах, присоединившихся к Союзу, процедура регистрации проводится на основании экспертного заключения уполномоченного органа (экспертной организации) референтного государства. При этом уполномоченным органом референтного государства выдается регистрационное удостоверение с указанием всех государств признания.
II. Процедуры регистрации и экспертизы медицинского изделия
16. Для регистрации медицинского изделия заявитель выбирает референтное государство и государства признания.
17. Заявитель представляет в уполномоченный орган (экспертную организацию) референтного государства следующие документы:
а) заявление на проведение экспертизы и регистрации медицинского изделия (на бумажном и (или) электронном носителях) по форме согласно приложениям N 2 и 3 (далее в настоящем разделе - заявление);
б) регистрационное досье на электронном носителе, содержащее документы по перечню согласно приложению N 4. В случае если законодательством государства-члена не предусмотрена возможность оформления указанных документов в электронном виде, уполномоченный орган (экспертная организация) референтного государства вправе запросить такие документы (их копии) на бумажном носителе. При этом документы, представленные на иностранном языке, должны иметь заверенный в установленном законодательством государства-члена порядке аутентичный перевод на русский язык;
в) копии документов, подтверждающих оплату экспертизы и регистрации медицинского изделия в референтном государстве.
18. Уполномоченным органом (экспертной организацией) референтного государства в течение 5 рабочих дней со дня поступления заявления и регистрационного досье проводится проверка полноты и достоверности содержащихся в них сведений, принимается решение о начале процедуры регистрации и экспертизы медицинского изделия и размещается заявление и регистрационное досье в своей информационной системе. Информация о медицинских изделиях, в отношении которых проводится процедура экспертизы и регистрации, и документы, содержащиеся в регистрационном досье, кроме инструкции по применению медицинского изделия и маркировки медицинского изделия, относятся к конфиденциальной информации и доступны только заинтересованным уполномоченным органам (экспертным организациям) государств-членов.
В случае если заявление представлено с нарушением требований, установленных настоящими Правилами, в заявлении указаны недостоверные сведения или регистрационное досье представлено не в полном объеме, уполномоченный орган (экспертная организация) референтного государства в течение 5 рабочих дней со дня поступления таких заявления и регистрационного досье уведомляет заявителя о необходимости устранения выявленных нарушений и (или) представления отсутствующих документов в срок, не превышающий 30 рабочих дней со дня размещения соответствующего уведомления в информационной системе уполномоченного органа (экспертной организации) референтного государства, путем передачи уведомления заявителю лично под расписку, либо направления уведомления заказным почтовым отправлением с уведомлением о вручении, либо передачи в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
В течение 3 рабочих дней со дня представления заявления и регистрационного досье, соответствующих требованиям настоящих Правил, уполномоченный орган (экспертная организация) референтного государства принимает решение о начале процедуры экспертизы и регистрации медицинского изделия.
19. Уполномоченные органы (экспертные организации) государств признания вправе ознакомиться с ходом проведения экспертных работ в референтом государстве, в том числе с перепиской заявителя и уполномоченного органа (экспертной организации) по вопросам устранения замечаний и с документами, представленными заявителем в процессе экспертизы и регистрации медицинских изделий.
20. Ответственность за достоверность предоставленного в уполномоченный орган (экспертную организацию) регистрационного досье несет заявитель.
21. При необходимости уполномоченный орган (экспертная организация) привлекает к участию в экспертизе экспертов, лиц, не работающих в уполномоченном органе (экспертной организации), если их специальные знания необходимы для проведения экспертизы.
Представители уполномоченных организаций, проводивших технические испытания, исследования (испытания) с целью оценки биологического действия и клинические испытания медицинского изделия, представленного на экспертизу, не могут привлекаться к участию в экспертизе.
При проведении экспертизы эксперт не может находиться в какой-либо зависимости от органа или лица, назначившего эту экспертизу, производителя медицинского изделия, его уполномоченного представителя или других заинтересованных в результатах экспертизы лиц.
В случае если эксперту известны обстоятельства, препятствующие его привлечению к проведению экспертизы либо не позволяющие ему соблюдать принципы ее проведения, он должен сообщить об этом руководителю уполномоченного органа (экспертной организации) референтного государства.
22. Уполномоченный орган (экспертная организация) референтного государства проводит экспертизу медицинского изделия и оформляет экспертное заключение согласно приложению N 5 в срок, не превышающий 60 рабочих дней со дня принятия им решения о начале процедуры регистрации и экспертизы медицинского изделия.
Выводы, содержащиеся в экспертном заключении, должны быть однозначными и понятными.
В случае если выводы экспертного заключения относительно возможности регистрации медицинского изделия в референтном государстве являются положительными, уполномоченный орган референтного государства в течение 5 рабочих дней со дня оформления экспертного заключения уведомляет заявителя о необходимости представления копий документов об оплате экспертизы и регистрации в государствах признания в срок, не превышающий 10 рабочих дней со дня размещения соответствующего уведомления в информационной системе уполномоченного органа (экспертной организации) референтного государства либо со дня получения уведомления заявителем лично под расписку, заказным почтовым отправлением с уведомлением о вручении, в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
23. Уполномоченный орган (экспертная организация) или организация, определенная уполномоченным органом (экспертной организацией) референтного государства, проводит инспекцию производства медицинских изделий в соответствии с требованиями, установленными Комиссией. Инспекция производства медицинских изделий проводится до подготовки экспертного заключения. Срок организации и проведения инспекции не входит в общий срок проведения экспертизы и не должен в совокупности превышать 90 рабочих дней.
24. Проведение экспертизы медицинского изделия включает в себя:
а) анализ документов и материалов, определяющих безопасность, эффективность и качество медицинского изделия, в том числе расходных материалов и комплектующих к медицинскому изделию;
б) анализ данных о разработке и производстве медицинского изделия (схемы процессов производства, основных стадий производства, упаковки, испытаний и процедуры выпуска конечного продукта);
в) анализ стандартов, которым соответствует медицинское изделие;
г) анализ протоколов технических испытаний (в части полноты проведения и компетентности испытательной лаборатории), а также признание результатов испытаний на основе этого анализа;
д) анализ отчетов по результатам инспекции производства медицинского изделия;
е) анализ отчетов по оценке биологического действия медицинского изделия (в части полноты и качества проведенных исследований, на соответствие правилам проведения испытаний (исследований) по оценке биологического действия медицинских изделий, утверждаемым Комиссией), а также признание результатов испытаний (исследований) на основе этого анализа;
ж) анализ и оценка клинических данных, содержащихся в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия, в том числе на соответствие клинических исследований правилам проведения клинических испытаний медицинских изделий, утверждаемым Комиссией, полноты проведенных исследований, достоверности результатов, сравнение клинических данных с имеющимися аналогами и признание результатов исследований на основе этого анализа;
з) анализ рисков (с указанием идентифицированных рисков, обобщенных данных по валидации и верификации испытаний, лабораторных тестов, подтверждающих возможность реализации научно-технических идей в конечном продукте, данные научной литературы по аналогам);
и) оценка соответствия указанного заявителем класса потенциального риска применения медицинского изделия в соответствии с правилами классификации медицинских изделий в зависимости от потенциального риска применения, утверждаемыми Комиссией;
к) анализ правильности определения номенклатурной принадлежности медицинского изделия согласно номенклатуре медицинских изделий, применяемой в Союзе;
л) анализ безопасности и эффективности лекарственного средства в составе медицинского изделия, его влияния на функциональность медицинского изделия, совместимости лекарственного средства с медицинским изделием (за исключением медицинских изделий для диагностики in vitro). Лекарственное средство должно быть зарегистрированным и разрешенным к применению в государстве - производителе лекарственного средства;
м) анализ биологической безопасности медицинского изделия на основе анализа всех материалов животного или человеческого происхождения, входящих в медицинское изделие, а также информации о подборе источников (доноров), отборе материала, процессинге, хранении, тестировании, валидации процедур тестирования, а также обращения с тканями, клетками, субстанциями животного или человеческого происхождения, культурами микроорганизмов и вирусов;
н) анализ процедуры и методов стерилизации медицинского изделия, материалов, обосновывающих способ стерилизации, предлагаемых методов контроля качества и определения остатков стерилизующего вещества при применении химического способа стерилизации;
о) изучение валидности программного обеспечения на основе анализа данных о его верификации и валидации, в том числе информации о его разработке и тестировании на предприятии и при мультицентровых исследованиях, данных об идентификации и маркировке операционной системы;
п) анализ отчета о стабильности медицинского изделия, обоснованности заявленного срока хранения;
р) анализ плана сбора данных по безопасности и эффективности медицинского изделия на постпродажном этапе;
с) анализ информации о маркетинге (если медицинское изделие находится в обращении на рынке более 2 лет) (при наличии);
т) анализ представленных производителем сведений о наличии или об отсутствии сообщений о несчастных случаях и отзывах с рынка медицинского изделия, о нежелательных событиях и (или) несчастных случаях, связанных с использованием медицинского изделия, уведомлений по безопасности медицинского изделия, подхода к рассмотрению этих проблем и их решения производителями в каждом из таких случаев, описания корректирующих действий, предпринятых в ответ на указанные случаи, а также соотношения уровня продаж и количества несчастных случаев и отзывов медицинского изделия из обращения;
у) анализ представленных производителем сведений о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них;
ф) оценка руководства пользователя (инструкции по медицинскому применению) и эксплуатационной документации;
х) оценка маркировки медицинского изделия;
ц) анализ документов, подтверждающих результаты испытаний медицинских изделий в целях утверждения типа средств измерений (в отношении медицинских изделий, отнесенных к средствам измерений, перечень которых утверждается Комиссией).
25. При проведении экспертизы медицинского изделия в случае недостаточности для подготовки экспертного заключения материалов и сведений, содержащихся в заявлении о регистрации и документах регистрационного досье, уполномоченный орган (экспертная организация) направляет заявителю соответствующий запрос с указанием характера замечаний и способа их устранения (далее - запрос). Запрос направляется однократно и может быть передан заявителю лично под расписку, направлен по почте заказным почтовым отправлением с уведомлением о вручении либо передан в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
Заявитель обязан представить ответ на запрос в срок, не превышающий 60 рабочих дней со дня получения запроса. В случае непредставления указанного ответа в срок уполномоченный орган (экспертная организация) принимает решение на основании документов, имеющихся в его распоряжении.
26. Период со дня направления запроса до дня получения уполномоченным органом (экспертной организацией) ответа на запрос не учитывается при исчислении срока проведения экспертизы медицинского изделия.
27. Основаниями для вынесения уполномоченным органом (экспертной организацией) заключения об отказе в регистрации медицинского изделия являются:
а) неподтверждение соответствующими материалами и сведениями, содержащимися в регистрационном досье, качества, и (или) эффективности, и (или) безопасности медицинского изделия;
б) превышение риска причинения вреда здоровью граждан и медицинских работников вследствие применения медицинского изделия над эффективностью его применения;
в) неустранение выявленных нарушений и (или) непредставление документов по запросу.
28. При необходимости уполномоченные органы (экспертные организации) государств признания могут направлять с использованием средств интегрированной системы Союза в уполномоченный орган (экспертную организацию) референтного государства свои замечания и предложения до оформления этим уполномоченным органом (экспертной организацией) экспертного заключения.
В ходе согласования экспертного заключения уполномоченные органы (экспертные организации) государств-членов могут взаимодействовать друг с другом для урегулирования возникающих вопросов.
29. После оформления экспертного заключения уполномоченный орган (экспертная организация) референтного государства размещает в своей информационной системе экспертное заключение. Уполномоченные органы (экспертные организации) государств признания в срок, не превышающий 30 календарных дней со дня размещения уполномоченным органом (экспертной организацией) референтного государства экспертного заключения, направляют в уполномоченный орган (экспертную организацию) референтного государства подтверждение согласования (несогласования) экспертного заключения (с обоснованием) по форме согласно приложению N 6 с использованием средств интегрированной системы Союза, в том числе правильности перевода руководства пользователя (инструкции по медицинскому применению), маркировки медицинского изделия на государственные языки в соответствии с требованиями законодательства государств-членов.
В случае непредставления государствами признания подтверждения согласования (несогласования) экспертного заключения в течение 30 календарных дней со дня размещения уполномоченным органом (экспертной организацией) референтного государства экспертного заключения экспертное заключение считается согласованным.
В течение 10 рабочих дней со дня согласования экспертного заключения государствами признания уполномоченный орган референтного государства принимает решение о регистрации медицинского изделия и размещает в едином реестре медицинских изделий, зарегистрированных в рамках Союза, сведения о медицинском изделии, руководство пользователя (инструкцию по медицинскому применению) и изображение утвержденной маркировки медицинского изделия.
30. Уполномоченный орган референтного государства в течение 10 рабочих дней со дня принятия решения о регистрации медицинского изделия оформляет регистрационное удостоверение и приложение к нему либо уведомляет заявителя об отказе в регистрации медицинского изделия лично под расписку, направляет уведомление по почте заказным почтовым отправлением с уведомлением о вручении либо передает его в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
III. Процедура согласования экспертного заключения
31. При проведении согласования экспертного заключения государства признания проводят оценку экспертного заключения референтного государства на предмет полноты и достаточности данных, подтверждающих безопасность, качество и эффективность медицинского изделия.
32. Согласование экспертного заключения является основанием для принятия решения о регистрации медицинского изделия.
33. Основанием для несогласования экспертного заключения референтного государства является наличие свидетельств о том, что эффективность и (или) безопасность медицинского изделия не подтверждены сведениями, представленными в регистрационном досье, или о том, что риск причинения вреда здоровью граждан и медицинских работников вследствие применения медицинского изделия превышает эффективность его применения.
34. При отсутствии консенсуса по согласованию экспертного заключения урегулирование разногласий осуществляется путем обращения уполномоченного органа референтного государства в консультативный комитет по медицинским изделиям при Коллегии Комиссии (далее - консультативный комитет).
Уполномоченный орган (экспертная организация) референтного государства направляет в консультативный комитет заявление на бланке уполномоченного органа (экспертной организации) о необходимости рассмотрения разногласий с указанием общих сведений о предмете разногласий и сведений об итогах проведения переговоров и консультаций. К заявлению могут прилагаться любые материалы, обосновывающие позицию уполномоченного органа (экспертной организации) референтного государства по предмету разногласий.
После получения заявления и прилагаемых к нему материалов от уполномоченного органа (экспертной организации) референтного государства консультативный комитет запрашивает у уполномоченных органов (экспертных организаций) государств признания материалы, подтверждающие их позицию по предмету разногласий.
После получения материалов от уполномоченных органов (экспертных организаций) консультативный комитет направляет уведомления о проведении заседания по урегулированию разногласий уполномоченным органам (экспертным организациям).
Консультативный комитет обеспечивает организацию и проведение заседания по урегулированию разногласий. В заседании принимают участие представители уполномоченных органов (экспертных организаций) референтного государства и государств признания.
По итогам заседания принимается решение, которое носит рекомендательный характер.
Срок урегулирования разногласий в отношении согласования экспертного отчета не должен превышать 30 рабочих дней со дня направления уполномоченным органом (экспертной организацией) референтного государства соответствующего заявления в консультативный комитет.
35. Несогласование экспертного заключения референтного государства в одном из государств признания является основанием для отказа в обращении медицинского изделия на территории этого государства.
IV. Экспертиза изменений, вносимых в регистрационное досье
36. Экспертиза изменений, вносимых в регистрационное досье, осуществляется уполномоченным органом (экспертной организацией) референтного государства и включает в себя оценку полноты, комплектности и правильности оформления документов, влияния вносимых изменений на безопасность, качество и эффективность медицинских изделий.
37. Производитель в течение 2 месяцев со дня внесения изменений в документы представленного в рамках регистрации медицинского изделия регистрационного досье обязан инициировать процедуру внесения изменений в регистрационное досье путем направления в уполномоченный орган (экспертную организацию) референтного государства соответствующего заявления по форме согласно приложению N 7 (далее в настоящем разделе - заявление) с приложением документов, подтверждающих изменения по перечню согласно приложению N 8.
38. Заявление и документы, подтверждающие изменения, размещаются уполномоченным органом (экспертной организацией) референтного государства в своей информационной системе и доступны только заинтересованным уполномоченным органам (экспертным организациям) государств-членов.
39. Уполномоченные органы (экспертные организации) государств признания в течение 30 рабочих дней со дня размещения уполномоченным органом (экспертной организацией) референтного государства в своей информационной системе заявления и документов, подтверждающих изменения, могут с использованием средств интегрированной системы Союза направить в уполномоченный орган (экспертную организацию) референтного государства свои замечания и предложения до подготовки этим органом (организацией) экспертного заключения по форме согласно приложению N 9.
В течение 5 рабочих дней со дня поступления заявления и документов, подтверждающих изменения, уполномоченный орган (экспертная организация) референтного государства проводит проверку полноты и достоверности содержащихся в них сведений.
В случае если заявление оформлено с нарушением требований, установленных настоящими Правилами, и (или) в заявлении указаны недостоверные сведения либо документы, подтверждающие изменения, представлены заявителем не в полном объеме, уполномоченный орган (экспертная организация) референтного государства не позднее 30 рабочих дней со дня поступления таких заявления и документов уведомляет заявителя о необходимости устранения выявленных нарушений и (или) представления отсутствующих документов путем передачи уведомления заявителю лично под расписку, либо направления уведомления заказным почтовым отправлением с уведомлением о вручении, либо передачи в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
В течение 3 рабочих дней со дня представления заявления и документов, подтверждающих изменения, оформленных надлежащим образом, уполномоченный орган (экспертная организация) референтного государства принимает решение о начале процедуры внесения изменений в регистрационное досье.
40. Заявитель представляет ответ на запрос уполномоченного органа (экспертной организации) в срок, не превышающий 60 календарных дней со дня получения этого запроса. В случае непредставления ответа в указанный срок уполномоченный орган (экспертная организация) принимает решение на основании документов, имеющихся в его распоряжении.
41. Период со дня направления уполномоченным органом (экспертной организацией) запроса до дня получения ответа на запрос не учитывается при исчислении срока проведения экспертизы медицинского изделия.
42. Внесение изменений в регистрационное досье осуществляется на основании результатов экспертизы этих изменений в срок, не превышающий 30 рабочих дней со дня представления заявления и документов, подтверждающих изменения, оформленных надлежащим образом.
43. Основаниями для подготовки уполномоченным органом (экспертной организацией) референтного государства экспертного заключения о невозможности внесения изменений в регистрационное досье являются:
а) недостоверность представленных сведений, обосновывающих внесение изменений;
б) отсутствие сведений, подтверждающих неизменность функционального назначения и (или) принципа действия медицинского изделия в связи с вносимыми изменениями;
в) неустранение выявленных нарушений и (или) непредставление отсутствующих документов.
44. Экспертное заключение и руководство пользователя (инструкция по медицинскому применению), изображение маркировки медицинского изделия на русском языке размещаются уполномоченным органом (экспертной организацией) референтного государства в своей информационной системе и доступны только заинтересованным уполномоченным органам (экспертным организациям) государств-членов.
45. Государства признания в течение 10 рабочих дней со дня размещения уполномоченным органом (экспертной организацией) референтного государства в своей информационной системе экспертного заключения вправе направить в уполномоченный орган (экспертную организацию) референтного государства замечания и предложения (с обоснованием).
46. В случае если вносимые изменения касаются сведений, содержащихся в регистрационном удостоверении, уполномоченный орган референтного государства выдает новое регистрационное удостоверение с сохранением прежнего номера (с указанием даты внесения соответствующих изменений).
47. Уполномоченный орган референтного государства в течение 10 рабочих дней со дня принятия соответствующего решения:
а) оформляет регистрационное удостоверение;
б) размещает в едином реестре медицинских изделий, зарегистрированных в рамках Союза, сведения о внесении изменений в регистрационное досье в порядке, установленном Комиссией, а также сканированные копии документов, в которые внесены изменения;
в) уведомляет заявителя об отказе во внесении изменений в регистрационное досье путем передачи уведомления лично под расписку, либо направления соответствующего уведомления заказным почтовым отправлением с уведомлением о вручении, либо передачи в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
V. Порядок приостановления или отмены действия
(аннулирования) регистрационного удостоверения
48. Приостановление действия регистрационного удостоверения осуществляется уполномоченным органом референтного государства в следующих случаях:
а) по результатам мониторинга безопасности, качества и эффективности медицинских изделий в пострегистрационный период - при выявлении потенциального серьезного риска для общественного здоровья;
б) по результатам государственного контроля за обращением медицинских изделий - при наличии сведений о фактах и обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий.
49. Решение о приостановлении действия регистрационного удостоверения (с указанием причин, даты и срока приостановления) принимается уполномоченным органом референтного государства в соответствии с законодательством этого государства-члена.
50. Срок приостановления действия регистрационного удостоверения не может превышать 6 месяцев, при этом реализация и применение таких медицинских изделий в рамках Союза не допускаются.
Уполномоченный орган референтного государства незамедлительно информирует уполномоченные органы государств признания, производителя или его уполномоченного представителя и Комиссию о приостановлении действия регистрационного удостоверения и вносит соответствующие сведения в единый реестр медицинских изделий, зарегистрированных в рамках Союза.
51. Заявитель в течение установленного уполномоченным органом референтного государства срока обязан устранить обстоятельства, повлекшие приостановление действия регистрационного удостоверения, уведомить об этом в письменной форме этот уполномоченный орган (с приложением подтверждающих документов). По результатам рассмотрения представленных заявителем документов уполномоченный орган референтного государства принимает решение о возобновлении либо об отмене действия (аннулировании) регистрационного удостоверения (с указанием даты возобновления, отмены действия (аннулирования) регистрационного удостоверения).
52. Уполномоченный орган референтного государства уведомляет заявителя о возобновлении действия регистрационного удостоверения в течение 5 рабочих дней со дня принятия такого решения лично под расписку, либо направляет уведомление заказным почтовым отправлением с уведомлением о вручении, либо передает в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью.
Решение о возобновлении действия регистрационного удостоверения принимается в соответствии с законодательством референтного государства и вступает в силу со дня его принятия.
53. В случае неустранения заявителем обстоятельств, повлекших приостановление действия регистрационного удостоверения, уполномоченный орган референтного государства принимает решение об отмене его действия (его аннулировании) (с обоснованием).
Уполномоченный орган референтного государства незамедлительно уведомляет заявителя об отмене действия (аннулировании) регистрационного удостоверения путем направления уведомления заявителю лично под расписку, либо направления уведомления заказным почтовым отправлением с уведомлением о вручении, либо передачи в электронной форме по телекоммуникационным каналам связи или в форме электронного документа, подписанного электронной подписью, и вносит соответствующие сведения в единый реестр медицинских изделий, зарегистрированных в рамках Союза.
Решение об отмене действия (аннулировании) регистрационного удостоверения принимается уполномоченным органом референтного государства также в случае подачи производителем или его уполномоченным представителем заявления об отмене действия регистрационного удостоверения по форме согласно приложению N 10.
54. Основаниями для принятия уполномоченным органом референтного государства решения об отмене действия (аннулировании) регистрационного удостоверения являются:
а) заявление производителя или его уполномоченного представителя об отмене действия (аннулировании) регистрационного удостоверения;
б) выявление случаев представления заявителем недостоверных сведений, которые не могли быть установлены при регистрации медицинского изделия;
в) вступившее в законную силу решение суда государства-члена;
г) представление уполномоченным органом государства-члена по результатам государственного контроля за обращением медицинских изделий сведений о фактах и обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинского изделия;
д) утрата медицинским изделием статуса медицинского в связи с внесением изменений в акты, составляющие право Союза.
VI. Процедура выдачи дубликата
регистрационного удостоверения
55. В случае утраты (порчи) регистрационного удостоверения заявитель вправе обратиться в уполномоченный орган референтного государства с заявлением о выдаче дубликата регистрационного удостоверения по форме согласно приложению N 11.
56. В случае порчи регистрационного удостоверения к заявлению о выдаче его дубликата прилагается испорченное регистрационное удостоверение.
57. В течение 7 рабочих дней со дня получения заявления о выдаче дубликата регистрационного удостоверения уполномоченный орган референтного государства оформляет дубликат регистрационного удостоверения на бланке регистрационного удостоверения и выдает его заявителю или направляет его заказным почтовым отправлением с уведомлением о вручении.
Приложение N 1
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА РЕГИСТРАЦИОННОГО УДОСТОВЕРЕНИЯ
МЕДИЦИНСКОГО ИЗДЕЛИЯ И ПРАВИЛА ЕГО ОФОРМЛЕНИЯ
I. Форма регистрационного удостоверения
медицинского изделия
Эмблема Евразийского экономического союза (1)
ЕВРАЗИЙСКИЙ ЭКОНОМИЧЕСКИЙ СОЮЗ (2)
___________________________________________________________________________
(полное наименование уполномоченного органа референтного государства) (3)
РЕГИСТРАЦИОННОЕ УДОСТОВЕРЕНИЕ (4)
МИ-XX-N ________ (5)
В соответствии с ______________________________________________________ (6)
(номер и дата приказа уполномоченного органа
референтного государства)
настоящее регистрационное удостоверение выдано: _______________________
_______________________________________________________________________ (7)
(полное наименование и страна производителя, включая место нахождения
(адрес) юридического лица)
_______________________________________________________________________ (8)
(полные наименования производственных площадок, включая место
нахождения (адрес) юридического лица)
_______________________________________________________________________ (9)
(наименование уполномоченного представителя производителя
на территориях государств - членов Евразийского экономического союза,
включая место нахождения (адрес) юридического лица)
в том, что ___________________________________________________________ (10)
(полное наименование медицинского изделия)
класс потенциального риска применения медицинского изделия: __________ (11)
вид медицинского изделия в соответствии с применяемой в Евразийском
экономическом союзе номенклатурой медицинских изделий ________________ (12)
зарегистрировано и разрешено к выпуску в обращение в рамках
Евразийского экономического союза
______________________________________________________________________ (13)
(полное наименование государства - члена Евразийского
экономического союза)
Перечень комплектующих, принадлежностей и расходных материалов
к модификации медицинского изделия приведен в приложении к
настоящему регистрационному удостоверению на __ л. (14)
Приложение является неотъемлемой частью настоящего регистрационного
удостоверения (15)
Срок действия регистрационного удостоверения: бессрочно (16)
Дата регистрации: "__" ______ 20__ г. (17)
Дата внесения изменений: "__" ______ 20__ г. (18)
______________________________________________________________________
(Ф. И. О. руководителя (уполномоченного лица)
уполномоченного органа)
_____________ М.П. (19)
(подпись)
N ______ (20)
Приложение к регистрационному удостоверению
МИ-XX-N ________ (1)
|
N п/п
|
Наименование составных частей медицинского изделия
|
|
1.
|
Основные блоки (части) медицинского изделия
|
|
2.
|
Принадлежности (при наличии)
|
|
3.
|
Расходные материалы (при наличии)
|
|
4.
|
Комплектующие (при наличии)
|
(2)
______________________________________________________________________
(Ф. И. О. руководителя (уполномоченного лица) уполномоченного органа)
___________ М.П. (3)
(подпись)
"__" __________ 20__ г.
II. Правила оформления регистрационного удостоверения
медицинского изделия
1. Регистрационное удостоверение заполняется уполномоченным органом государства - члена Евразийского экономического союза, осуществляющим регистрацию медицинского изделия, на русском языке с использованием электронных печатающих устройств и в случае наличия соответствующего требования в законодательстве референтного государства на государственном языке этого государства.
2. Заполнение регистрационного удостоверения на русском языке и государственном языке государства признания осуществляется на разных сторонах регистрационного удостоверения.
3. Регистрационное удостоверение относится к документам строгой отчетности, бланки изготавливаются типографским способом.
При необходимости наименование производителя, его место нахождения (адрес юридического лица), фактический адрес (кроме наименования государства) и сведения о продукции (тип, марка, модель, артикул и др.) могут быть указаны с использованием букв латинского алфавита.
4. Все поля регистрационного удостоверения должны быть заполнены (в оригинале регистрационного удостоверения нумерация полей отсутствует).
5. В регистрационном удостоверении указываются:
а) в поле 1 - эмблема Евразийского экономического союза;
б) в поле 2 - надпись, выполненная в 1 строку:
"ЕВРАЗИЙСКИЙ ЭКОНОМИЧЕСКИЙ СОЮЗ";
в) в поле 3 - полное наименование уполномоченного органа референтного государства;
г) в поле 4 - надпись, выполненная в одну строку "Регистрационное удостоверение";
д) в поле 5 - регистрационный номер регистрационного удостоверения и дата его выдачи.
Регистрационный номер регистрационного удостоверения формируется в следующем порядке:
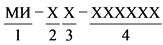 ,
,
где:
элемент 1 - медицинское изделие;
элемент 2 - 2-значный буквенный код референтного государства в соответствии с классификатором стран мира;
элемент 3 - 2-значный буквенный код государств признания в соответствии с классификатором стран мира (указываются коды всех государств признания, подтвердивших согласование экспертного заключения референтного государства);
элемент 4 - 6-значный порядковый номер регистрационного удостоверения, присвоенный уполномоченным органом референтного государства (присваивается автоматически из единого реестра медицинских изделий, зарегистрированных в рамках Евразийского экономического союза);
е) в поле 6 - номер и дата приказа уполномоченного органа референтного государства;
ж) в поле 7 - полное наименование и страна производителя, место нахождения (адрес юридического лица), фактический адрес - для юридического лица и его филиалов, которые производят продукцию, или фамилия, имя, отчество (при наличии), место жительства - для физического лица, зарегистрированного в качестве индивидуального предпринимателя;
з) в поле 8 - наименования производственных площадок, выпускающих медицинское изделие, место нахождения (адрес юридического лица), фактический адрес - для юридического лица и его филиалов, которые производят продукцию, или фамилия, имя, отчество (при наличии), место жительства - для физического лица, зарегистрированного в качестве индивидуального предпринимателя;
и) в поле 9 - наименование уполномоченного представителя производителя на территории государства-члена, его место нахождения (адрес юридического лица), фактический адрес - для юридического лица и его филиалов, которые производят продукцию, или фамилия, имя, отчество (при наличии), место жительства - для физического лица, зарегистрированного в качестве индивидуального предпринимателя;
к) в поле 10 - полное наименование медицинского изделия, которое должно соответствовать наименованию, указанному в экспертном заключении уполномоченного органа референтного государства, торговое название медицинского изделия (при наличии), сведения о медицинском изделии, обеспечивающие его идентификацию (тип, марка, модель, артикул и др.);
л) в поле 11 - класс потенциального риска применения медицинского изделия, подтвержденный при проведении экспертизы медицинского изделия в референтном государстве;
м) в поле 12 - вид медицинского изделия, в соответствии с применяемой в Евразийском экономическом союзе номенклатурой медицинских изделий;
н) в поле 13 - наименования референтного государства и государств признания;
о) в поле 14 - количество листов приложения к регистрационному удостоверению (заполняется при наличии приложения);
п) в поле 17 - дата регистрации медицинского изделия, которая указывается словесно-цифровым способом: число - двумя арабскими цифрами (в кавычках), месяц - словом, год - четырьмя арабскими цифрами (с указанием сокращенного обозначения года "г.)";
р) в поле 18 - дата внесения изменений в регистрационное удостоверение, которая указывается словесно-цифровым способом: число - двумя арабскими цифрами (в кавычках), месяц - словом, год - четырьмя арабскими цифрами (с указанием сокращенного обозначения года "г."). Данное поле заполняется при внесении изменений в регистрационное досье с выдачей нового регистрационного удостоверения с прежним номером;
с) в поле 19 - должность, подпись, фамилия, имя, отчество (при наличии) руководителя (уполномоченного лица) уполномоченного органа, выдавшего свидетельство, заверенные печатью этого уполномоченного органа. Использование факсимиле вместо подписи не допускается;
т) в поле 20 - типографский номер, серия и порядковый номер бланка регистрационного удостоверения, проставляемый при его изготовлении.
5. При наличии составных частей медицинского изделия, включающих основные блоки (части) медицинского изделия, комплектующих, принадлежностей и расходных материалов к медицинскому изделию заполняется приложение к регистрационному удостоверению, которое является неотъемлемой частью регистрационного удостоверения. Каждый лист приложения должен быть пронумерован. В приложении к регистрационному удостоверению указываются:
а) в поле 1 - регистрационный номер регистрационного удостоверения и дата его выдачи;
б) в поле 2 - перечень составных частей медицинского изделия, включающих основные блоки (части) медицинского изделия, комплектующих, принадлежностей и расходных материалов к медицинскому изделию, указание модели (при наличии);
в) в поле 3 - должность, подпись, фамилия, имя, отчество (при наличии) руководителя (уполномоченного лица) уполномоченного органа, выдавшего свидетельство, заверенные печатью этого уполномоченного органа. Использование факсимиле вместо подписи не допускается.
6. При заполнении регистрационного удостоверения и (или) приложения к нему указание сведений, не предусмотренных настоящими правилами, а также использование сокращений слов (кроме общепринятых) и исправление текста не допускаются.
7. В случае утраты или порчи регистрационного удостоверения уполномоченным органом референтного государства выдается дубликат этого регистрационного удостоверения. При этом в правом верхнем углу регистрационного удостоверения проставляются пометки: "Дубликат выдан "__" __________ 20__ г." и "Оригинал регистрационного удостоверения признается недействующим".
Приложение N 2
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАЯВЛЕНИЯ
О ПРОВЕДЕНИИ ЭКСПЕРТИЗЫ МЕДИЦИНСКОГО ИЗДЕЛИЯ
В уполномоченный орган
(экспертную организацию)
государства - члена
Евразийского экономического союза
_________________________________
(наименование референтного
государства)
_________________________________
(наименование государства
признания)
ЗАЯВЛЕНИЕ
о проведении экспертизы медицинского изделия
___________________________________________________________________________
(полное и сокращенное (при наличии), в том числе фирменное, наименования
организации, от имени которой производится регистрация (производитель
(уполномоченный представитель производителя), организационно-правовая
форма юридического лица)
настоящим просит произвести экспертизу медицинского изделия
в качестве ________________________________________________________________
(референтного государства, государства признания -
указать нужное)
|
1.
|
Наименование медицинского изделия
|
|||||||||||||||
|
2.
|
Назначение медицинского изделия
|
|||||||||||||||
|
3.
|
Область применения медицинского изделия
|
|||||||||||||||
|
4.
|
Класс потенциального риска применения медицинского изделия
|
|||||||||||||||
|
5.
|
Код вида медицинского изделия (согласно применяемой в Союзе номенклатуре медицинских изделий)
|
|||||||||||||||
|
6.
|
В составе медицинского изделия имеется лекарственное средство (выделить нужное)
|
|
Да
|
|||||||||||||
|
|
Нет
|
|||||||||||||||
|
7.
|
Комплектация медицинского изделия
|
|||||||||||||||
|
N
|
Наименование
|
Модель
|
Производитель
|
Страна
|
||||||||||||
|
1) Основной блок
(при наличии)
|
||||||||||||||||
|
2) Комплектующие
(при наличии)
|
||||||||||||||||
|
3) Расходные материалы
(при наличии)
|
||||||||||||||||
|
4) Принадлежности
(при наличии)
|
||||||||||||||||
|
8.
|
Срок хранения/гарантийный срок эксплуатации
|
|||||||||||||||
|
9.
|
Условия хранения
|
|||||||||||||||
|
10.
|
Регистрация в стране-производителе и других странах
|
|||||||||||||||
|
1.
|
Наименование страны
|
N регистрационного удостоверения
(при наличии)
|
Дата выдачи
|
Срок действия
|
||||||||||||
|
2.
|
||||||||||||||||
|
...
|
||||||||||||||||
|
11.
|
Производство
|
полностью на данном производстве
частично на данном производстве
полностью на другом производстве
|
||||||||||||||
|
12.
|
Сведения о производителе
|
|||||||||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа
|
юридический адрес
|
фактический адрес
|
номера телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
|||||||||
|
13.
|
Сведения о производственной(ых) площадке(ах)
|
|||||||||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа (при наличии)
|
фактический адрес
|
номера телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
||||||||||
|
14.
|
Сведения об уполномоченном представителе (при наличии)
|
|||||||||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа (при наличии)
|
юридический адрес
|
фактический адрес
|
номера телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
|||||||||
|
15.
|
Сведения о документе, подтверждающем оплату за проведение экспертизы медицинского изделия
|
|||||||||||||||
|
Гарантирую достоверность и идентичность информации, содержащейся в регистрационном досье и заявлении.
|
||||||||||||||||
|
Дата подачи заявления
|
||||||||||||||||
|
Ф. И. О. и должность руководителя производителя (уполномоченного представителя)
|
||||||||||||||||
|
Подпись, печать производителя (уполномоченного представителя)
|
||||||||||||||||
Приложение N 3
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАЯВЛЕНИЯ
О ПРОВЕДЕНИИ РЕГИСТРАЦИИ МЕДИЦИНСКОГО ИЗДЕЛИЯ
В уполномоченный орган
государства - члена
Евразийского экономического союза
_________________________________
(наименование референтного
государства)
_________________________________
(наименование государства
признания)
ЗАЯВЛЕНИЕ
о проведении регистрации медицинского изделия
___________________________________________________________________________
(полное и сокращенное (при наличии), в том числе фирменное, наименования
организации, от имени которой производится регистрация (производитель
(уполномоченный представитель производителя), организационно-правовая
форма юридического лица)
настоящим просит произвести регистрацию медицинского изделия
в качестве ________________________________________________________________
(референтного государства, государства признания -
указать нужное)
|
1.
|
Наименование медицинского изделия
|
|||||||||
|
2.
|
Назначение медицинского изделия
|
|||||||||
|
3.
|
Область применения медицинского изделия
|
|||||||||
|
4.
|
Класс потенциального риска применения медицинского изделия
|
|||||||||
|
5.
|
Код вида медицинского изделия (согласно применяемой в Союзе номенклатуре медицинских изделий)
|
|||||||||
|
6.
|
В составе медицинского изделия имеется лекарственное средство (выделить нужное)
|
|
Да
|
|||||||
|
|
Нет
|
|||||||||
|
7.
|
Перечень комплектующих
|
|||||||||
|
8.
|
Сведения о производителе
|
|||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа
|
юридический адрес
|
фактический адрес
|
номера телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
|||
|
9.
|
Сведения о производственной(ых) площадке(ах)
|
|||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа (при наличии)
|
фактический адрес
|
номер телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
||||
|
10.
|
Сведения об уполномоченном представителе (при наличии)
|
|||||||||
|
N
|
Наименование, страна
|
номер, дата и срок действия разрешительного документа (при наличии)
|
юридический адрес
|
фактический адрес
|
номера телефона и факса, адрес электронной почты (при наличии)
|
Ф. И. О. и должность руководителя
|
Ф. И. О. и должность контактного лица
|
|||
|
11.
|
Сведения о документе, подтверждающем оплату государственной пошлины за проведение регистрации медицинского изделия
|
|||||||||
|
Гарантирую достоверность и идентичность информации, содержащейся в регистрационном досье и заявлении.
|
||||||||||
|
Дата подачи заявления
|
||||||||||
|
Ф. И. О. и должность руководителя производителя (уполномоченного представителя)
|
||||||||||
|
Подпись, печать производителя (уполномоченного представителя)
|
||||||||||
Приложение N 4
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ПЕРЕЧЕНЬ
ДОКУМЕНТОВ, НЕОБХОДИМЫХ ДЛЯ РЕГИСТРАЦИИ МЕДИЦИНСКОГО
ИЗДЕЛИЯ, И ФОРМА СПРАВКИ НА МЕДИЦИНСКОЕ ИЗДЕЛИЕ
|
N п/п
|
Наименование документа
|
Медицинское изделие класса
|
Медицинское изделие для диагностики
in vitro
(независимо от класса потенциального риска применения)
|
Примечание
|
|||
|
1
|
2а
|
2б
|
3
|
||||
|
1.
|
Заявление
|
+
|
+
|
+
|
+
|
+
|
по формам, предусмотренным приложениями N 2 и 3 к Правилам
|
|
2.
|
Доверенность от производителя на право представления интересов при регистрации (при необходимости)
|
+
|
+
|
+
|
+
|
+
|
в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства - члена Евразийского экономического союза (далее - государство-член)
|
|
3.
|
Копия разрешительного документа на право производства в стране-производителе с приложением (при наличии)
|
+
|
+
|
+
|
+
|
+
|
в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена
|
|
4.
|
Копии сертификатов на систему менеджмента качества производителя медицинских изделий (ИСО 13485 либо соответствующий региональный или национальный стандарт государства-члена) (при наличии)
|
+
|
+
|
+
|
+
|
+
|
в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена
|
|
5.
|
Декларация о соответствии требованиям безопасности и эффективности медицинских изделий или эквивалентный документ (при наличии)
|
+
|
+
|
+
|
+
|
+
|
|
|
6.
|
Копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт (за исключением медицинских изделий, впервые произведенных на территории государства-члена)), выданного в стране производителя (при наличии) с представлением перевода на русский язык
|
+
|
+
|
+
|
+
|
+
|
в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством государства-члена
|
|
7.
|
Копия документа, удостоверяющего регистрацию в других странах (при наличии)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
8.
|
Справка на медицинское изделие с описанием области применения, назначения, краткой характеристики медицинского изделия, вариантами исполнения и комплектующими (по форме)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
9.
|
Данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
10.
|
Информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
11.
|
Сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
12.
|
Информация о маркетинге (история при условии обращения изделия на рынке более 2 лет) (при наличии)
|
-
|
-
|
+
|
+
|
+
(кроме 1 и 2а классов)
|
заверяется производителем (его уполномоченным представителем)
|
|
13.
|
Сообщения о несчастных случаях и отзывах (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий):
список нежелательных событий или несчастных случаев, связанных с использованием изделия, и указание периода времени, на протяжении которого происходили указанные случаи
если нежелательных событий слишком много, необходимо предоставить краткие обзоры по каждому из типов событий и указать общее количество событий каждого типа, о которых поступали отчеты
список отзывов с рынка медицинских изделий и (или) пояснительных уведомлений и описание подхода к рассмотрению этих проблем и их решению производителями в каждом из таких случаев
описание анализа и (или) корректирующих действий, предпринятых в ответ на указанные случаи
|
+
|
+
|
+
|
+
|
+
(кроме 1 класса)
|
заверяется производителем (его уполномоченным представителем)
|
|
14.
|
Перечень стандартов, которым соответствует медицинское изделие (с указанием сведений о них)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
15.
|
Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (далее - общие требования)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
16.
|
Документ, устанавливающий требования к техническим характеристикам медицинского изделия
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
17.
|
Протоколы технических испытаний, проведенных в целях доказательства соответствия общим требованиям
|
+
|
+
|
+
|
+
|
+
(за исключением реагентов, наборов реагентов)
|
|
|
18.
|
Протоколы исследований (испытаний) по оценке биологического действия медицинского изделия, проведенных в целях доказательства соответствия общим требованиям
|
+
|
+
|
+
|
+
|
-
|
|
|
19.
|
Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия
|
+
|
+
|
+
|
+
|
+
(кроме 1 класса)
|
заверяется производителем (его уполномоченным представителем)
|
|
20.
|
Отчет об анализе рисков
|
-
|
+
|
+
|
+
|
+
(кроме 1 класса)
|
заверяется производителем (его уполномоченным представителем)
|
|
21.
|
Данные о лекарственных средствах в составе медицинского изделия (состав лекарственного средства, количество, данные о совместимости лекарственного средства с медицинским изделием, регистрации лекарственного средства в стране-производителе)
|
+
|
+
|
+
|
+
|
-
|
заверяется производителем (его уполномоченным представителем)
|
|
22.
|
Данные о биологической безопасности (при наличии)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
23.
|
Данные о процедуре стерилизации, включая информацию о валидации процесса, результаты тестирования на содержание микроорганизмов (степень биологической нагрузки), пирогенности, стерильности (при необходимости) с указанием методов проведения испытаний и данные о валидации упаковки (для стерильных изделий)
|
+
|
+
|
+
|
+
|
+
(кроме 1 класса)
|
заверяется производителем (его уполномоченным представителем)
|
|
24.
|
Информация о специальном программном обеспечении (при наличии): сведения производителя о валидации программного обеспечения
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
25.
|
Отчет об исследованиях стабильности - с аутентичным переводом на русский язык результатов и выводов испытаний для изделий, имеющих срок хранения
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
26.
|
Эксплуатационный документ или инструкция по применению медицинского изделия на государственном языке государств признания (при необходимости) и русском языках
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
27.
|
Руководство по сервисному обслуживанию (в части комплектующих медицинского изделия) - в случае отсутствия данных в эксплуатационной документации (при наличии)
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
28.
|
Отчет об инспекции производства (при наличии)
|
+
|
+
|
+
|
+
|
+
|
|
|
29.
|
План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе
|
+
|
+
|
+
|
+
|
+
|
заверяется производителем (его уполномоченным представителем)
|
|
30.
|
Документы, подтверждающие результаты испытаний медицинских изделий в целях утверждения типа средств измерений (в отношении медицинских изделий, отнесенных к средствам измерений, перечень которых утверждается Комиссией) (при необходимости)
|
+
|
+
|
+
|
+
|
+
|
|
(форма)
Справка на медицинское изделие
|
Наименование
|
Производитель (страна)
|
Комплектность
|
Область применения, назначение
|
Краткая характеристика медицинского изделия
|
|||
|
наименование составных частей
|
модель
|
производитель
|
страна
|
||||
|
1. Основные блоки (части) медицинского изделия
|
|||||||
|
2. Принадлежности
(при наличии)
|
|||||||
|
3. Расходные материалы
(при наличии)
|
|||||||
|
4. Комплектующие
(при наличии)
|
|||||||
Приложение N 5
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ЭКСПЕРТНОЕ ЗАКЛЮЧЕНИЕ
об оценке безопасности, эффективности и качества
медицинских изделий при регистрации
1. Спецификация на медицинское изделие:
а) наименование;
б) производитель, страна;
в) производственная площадка, страна;
г) область применения и назначение;
д) вид медицинского изделия в соответствии с номенклатурой медицинских изделий, применяемой в Евразийском экономическом союзе;
е) класс потенциального риска применения медицинского изделия;
ж) описание комплектующих и расходных материалов;
з) описание или список модификаций медицинского изделия;
и) состав медицинского изделия;
к) материалы, из которых изготовлено медицинское изделие;
л) основные технические характеристики.
2. Данные о сертификации системы менеджмента качества (при наличии):
система менеджмента качества производителя сертифицирована на соответствие требованиям:
___________________________________________________________________________
(перечисление сертификатов с описанием идентификационных данных:
номера, даты выдачи, срока действия, наименования органа по сертификации)
3. Разработка и производство:
а) оценка представленных заявителем данных о разработке и производстве, включая анализ отчета инспектирования производства (при наличии);
б) заключение о соответствии разработки, технологического процесса и контроля качества производству безопасной и качественной продукции.
4. Используемые стандарты, которые применяют производители медицинских изделий, в том числе на материалы изготовления, комплектующие, расходные материалы, методы тестирования, стандарты, использованные при проведении технических испытаний, исследований (испытаний) по оценке биологического действия, клинических испытаний медицинского изделия.
5. Обоснованное заключение о соответствии используемых стандартов общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (далее - общие требования).
6. Документ, устанавливающий требования к техническим характеристикам медицинского изделия:
а) анализ документа на соответствие общим требованиям;
б) заключение о соответствии технических характеристик медицинского изделия общим требованиям.
7. Оценка технических испытаний и исследований (испытаний) с целью оценки биологического действия:
а) анализ протоколов технических испытаний и исследований (испытаний) с целью оценки биологического действия с точки зрения полноты и достоверности проведения испытаний в сравнении с требованиями спецификаций производителя или технических условий (при наличии);
б) заключение о полноте и достоверности проведенных технических испытаний и исследований (испытаний) с целью оценки биологического действия.
8. Анализ результатов клинических испытаний (исследований) или клинических данных:
а) анализ соответствия клинических испытаний (исследований) правилам проведения клинических испытаний (исследований) медицинских изделий, утверждаемым Комиссией, полноты и достоверности результатов проведенных испытаний;
б) заключение о соответствии клинических испытаний (исследований) правилам проведения клинических испытаний (исследований), утверждаемым Комиссией, полноты и достоверности их результатов, обоснованности выводов;
в) анализ отчета о клиническом доказательстве эффективности и безопасности медицинского изделия.
9. Анализ рисков: заключение о полноте и достоверности проведенного анализа рисков.
10. Медицинские изделия, содержащие лекарственные средства: заключение о безопасности и эффективности медицинского изделия, содержащего лекарственное средство, влиянии лекарственного средства на функциональность медицинского изделия и совместимости лекарственного средства с медицинским изделием.
11. Биологическая безопасность медицинского изделия: заключение о биологической безопасности медицинского изделия на основе анализа перечня всех материалов животного или человеческого происхождения, а также наночастиц, генно-модифицированных организмов и других вновь разрабатываемых материалов, включенных в изделие, информации о подборе источников (доноров), отбора материала, процессинга, хранения, тестирования, валидации процедур тестирования и обращения с тканями, клетками, субстанциями животного или человеческого происхождения, культурами микроорганизмов и вирусов.
12. Стерилизация: анализ процедуры стерилизации на основе изучения данных о разработке методов стерилизации медицинского изделия и материалов, обосновывающих способы стерилизации, предлагаемых методов контроля качества и определения остатков стерилизующего вещества при применении способа стерилизации.
13. Медицинские изделия, включающие в себя специальное программное обеспечение: заключение о валидности специального программного обеспечения на основе анализа данных о его верификации и валидации, в том числе информации о его разработке, тестировании внутри предприятия и при клинических многоцентровых исследованиях, данных об идентификации и маркировке операционной системы.
14. Анализ и заключение по данным о стабильности медицинского изделия.
15. Анализ плана сбора и анализа данных по безопасности и эффективности медицинского изделия на постпродажном этапе.
16. Анализ информации и заключение о маркетинге медицинского изделия (история при условии обращения изделия на рынке более 2 лет) (при наличии).
17. Анализ сообщений о несчастных случаях и отзывах о нежелательных событиях (несчастных случаях), связанных с использованием медицинского изделия, анализ случаев изъятия медицинских изделий из обращения и (или) пояснительных уведомлений, письменные доказательства об устранении обстоятельств изъятий медицинских изделий из обращения, описания корректирующих действий, предпринятых производителем в ответ на указанные случаи, соотношения уровня продаж к количеству несчастных случаев и отзывов (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий).
18. Анализ представленных производителем сведений о соответствии медицинского изделия общим требованиям:
а) заключение о правильности определения требований, относящихся к данному медицинскому изделию, полноте и достоверности доказательств соответствия общим требованиям.
19. Анализ отчетов об инспекции производства (при наличии).
20. Анализ документов, подтверждающих результаты испытаний медицинских изделий в целях утверждения типа средств измерений (в отношении медицинских изделий, отнесенных к средствам измерений, перечень которых утверждается Комиссией) (при необходимости).
21. Общее заключение о подтверждении (не подтверждении) безопасности, качества и эффективности медицинского изделия, рекомендация о возможности (невозможности) регистрации медицинского изделия.
22. Ф. И. О., должность, ученая степень (звание) (при наличии), подпись экспертов, проводивших экспертизу.
23. Дата составления отчета.
24. Подпись руководителя экспертной организации.
25. Печать экспертной организации.
Приложение N 6
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАКЛЮЧЕНИЯ
О ПОДТВЕРЖДЕНИИ СОГЛАСОВАНИЯ (НЕСОГЛАСОВАНИЯ)
ЭКСПЕРТНОГО ЗАКЛЮЧЕНИЯ ПО РЕЗУЛЬТАТАМ ЭКСПЕРТИЗЫ
БЕЗОПАСНОСТИ, ЭФФЕКТИВНОСТИ И КАЧЕСТВА МЕДИЦИНСКОГО ИЗДЕЛИЯ
ГОСУДАРСТВА - ЧЛЕНА ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА,
ОСУЩЕСТВЛЯЮЩЕГО РЕГИСТРАЦИЮ МЕДИЦИНСКОГО ИЗДЕЛИЯ
___________________________________________________________________________
(наименование уполномоченного органа (экспертной организации)
государства признания)
УТВЕРЖДАЮ
(руководитель уполномоченного органа
(экспертной организации)
Ф. И. О., подпись, печать)
"__" __________ 20__ г.
ЗАКЛЮЧЕНИЕ
о подтверждении согласования (несогласования) экспертного
заключения по результатам экспертизы безопасности,
эффективности и качества медицинского изделия
государства - члена Евразийского экономического союза,
осуществляющего регистрацию медицинского изделия
N ____ от "__" __________ 20__ г.
|
1. Наименование уполномоченного органа (экспертной организации) государства - члена Евразийского экономического союза, осуществляющего регистрацию медицинского изделия (далее - референтное государство)
|
||
|
2. Дата размещения экспертного заключения в информационной системе уполномоченного органа (экспертной организации) референтного государства
|
||
|
3. Номер экспертного заключения
|
||
|
4. Наименование медицинского изделия (с указанием принадлежностей, необходимых для применения медицинского изделия по назначению)
|
||
|
5. Производитель медицинского изделия (полное и сокращенное наименования, организационно-правовая форма юридического лица, местонахождение, адрес)
|
||
|
6. Уполномоченный представитель производителя (полное и сокращенное наименования, организационно-правовая форма юридического лица, местонахождение, адрес) (при наличии)
|
||
|
7. Заявитель
|
||
|
8. Сведения об экспертах (Ф. И. О., специальность, ученая степень (звание) (при наличии), место работы и должность)
|
||
|
9. Анализ представленного уполномоченным органом (экспертной организацией) референтного государства экспертного заключения по результатам экспертизы безопасности, качества и эффективности медицинского изделия на предмет полноты, качества экспертизы документов, оценки объема и полноты проведенных испытаний и исследований
|
||
|
11. Результаты экспертизы (указываются выводы по отдельным аспектам экспертного заключения)
|
||
|
12. Вывод (указывается общий вывод, а в случае вынесения отрицательного заключения - с обоснованием причин)
|
||
|
Дата направления заключения о подтверждении согласования (несогласования) экспертного заключения референтного государства по результатам экспертизы безопасности, эффективности и качества медицинского изделия
|
||
|
Об ответственности за достоверность сведений, изложенных в заключении о подтверждении согласования (несогласования) экспертного заключения референтного государства по результатам экспертизы безопасности, эффективности и качества медицинского изделия, предупрежден.
|
||
|
___________________________
(Ф. И. О. эксперта)
|
_________________________
(подпись)
|
|
Приложение N 7
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАЯВЛЕНИЯ
О ВНЕСЕНИИ ИЗМЕНЕНИЙ В РЕГИСТРАЦИОННОЕ ДОСЬЕ
МЕДИЦИНСКОГО ИЗДЕЛИЯ
В уполномоченный орган
государства - члена
Евразийского экономического союза
_________________________________
(наименование референтного
государства)
_________________________________
(наименование государства
признания)
ЗАЯВЛЕНИЕ
о внесении изменений в регистрационное досье медицинского изделия
___________________________________________________________________________
(полное и сокращенное (при наличии), в том числе фирменное, наименования
организации, от имени которой производится внесение изменений
(производитель (уполномоченный представитель производителя),
организационно-правовая форма юридического лица)
|
1.
|
Наименование медицинского изделия
|
||
|
2.
|
Производитель медицинского изделия
|
||
|
3.
|
Страна производства медицинского изделия
|
||
|
4.
|
Уполномоченный представитель производителя (при наличии)
|
||
|
5.
|
Класс потенциального риска применения медицинского изделия
|
||
|
5.
|
Код вида медицинского изделия (согласно применяемой в Союзе номенклатуре медицинских изделий)
|
||
настоящим просит произвести внесение изменений в регистрационное досье медицинского изделия в связи со следующими изменениями:
|
Изменения, вносимые в регистрационное досье
|
||
|
1.
|
Изменение сведений о заявителе, включая сведения о реорганизации юридического лица, об изменении его наименования или фамилии, имени и адреса места жительства индивидуального предпринимателя
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
|
2.
|
Изменение наименования медицинского изделия
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
|
3.
|
Изменение состава принадлежностей, и (или) комплектующих, и (или) расходных материалов
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
|
4.
|
Изменение показаний по применению, области применения, противопоказаний; побочных эффектов
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
|
5.
|
Изменение сведений о производителе медицинского изделия
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
|
6.
|
Изменение в технической и (или) эксплуатационной документации медицинского изделия
|
|
|
Редакция до внесения изменений
|
Вносимые изменения
|
|
Внесение изменений в регистрационное досье не влечет изменения свойств
и характеристик, влияющих на безопасность, качество и эффективность
медицинского изделия.
___________________________________________________________________________
(производитель медицинского изделия (его уполномоченный представитель))
Гарантирую достоверность представленной информации.
Гарантирую сохранение заявленных характеристик безопасности и
эффективности медицинского изделия в течение всего срока службы при
соблюдении условий эксплуатации, транспортирования и хранения медицинского
изделия в соответствии с требованиями завода-производителя.
Дата подачи заявления
Ф. И. О. и должность руководителя
производителя (уполномоченного
представителя)
Подпись, печать производителя
(уполномоченного представителя)
Приложение N 8
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ПЕРЕЧЕНЬ
ИЗМЕНЕНИЙ, ВНОСИМЫХ В РЕГИСТРАЦИОННОЕ ДОСЬЕ МЕДИЦИНСКОГО
ИЗДЕЛИЯ В ПЕРИОД ДЕЙСТВИЯ РЕГИСТРАЦИОННОГО УДОСТОВЕРЕНИЯ
И НЕ ТРЕБУЮЩИХ НОВОЙ РЕГИСТРАЦИИ
|
Наименование изменяемых сведений
|
Условия
|
Сведения и документы, необходимые для внесения изменений
|
|
1. Сведения о заявителе, включая сведения о реорганизации юридического лица, о изменении его наименования или фамилии, имени, адреса места жительства индивидуального предпринимателя
|
внесение изменений в регистрационное удостоверение не влияет на эффективность и безопасность медицинского изделия в соответствии с Общими требованиями безопасности и эффективности медицинских изделий, требованиями к их маркировке и эксплуатационной документации на них
|
копия документа, подтверждающего полномочия уполномоченного представителя производителя
|
|
номер регистрационного досье
|
||
|
документы, подтверждающие изменения
|
||
|
опись документов
|
||
|
2. Наименование медицинского изделия
|
мотивированное обоснование необходимости изменения наименования медицинского изделия, не влияющего на функциональные и технические характеристики
|
заявление о внесении изменений по форме, предусмотренной приложением N 7 к Правилам
|
|
документ <*>, удостоверяющий регистрацию медицинского изделия в стране производителе (декларация соответствия; регистрационное удостоверение, сертификат свободной продажи, сертификат на экспорт и др.) с внесенными изменениями
|
||
|
копия регистрационного удостоверения, оформленного по форме, предусмотренной приложением N 1 к Правилам
|
||
|
письмо производителя, содержащее мотивированное обоснование необходимости изменения наименования медицинского изделия, не влияющего на функциональные и технические характеристики медицинского изделия
|
||
|
проекты инструкций по применению (руководство пользователя) медицинского изделия
|
||
|
макет маркировки
|
||
|
документ, устанавливающий требования к техническим характеристикам медицинского изделия, которым соответствует медицинское изделие, приведенный в соответствие с новым наименованием медицинского изделия
|
||
|
эксплуатационная документация производителя на медицинское изделие, в том числе инструкция по применению (руководство по эксплуатации) медицинского изделия, приведенная в соответствие с новым наименованием медицинского изделия
|
||
|
фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 x 24 см)
|
||
|
опись документов
|
||
|
3. Состав принадлежностей, комплектующих и (или) расходных материалов
|
отсутствие влияния на функциональные характеристики медицинского изделий
|
заявление о внесении изменений по форме, предусмотренной приложением N 7 к Правилам
|
|
копия регистрационного удостоверения, оформленного по единой форме, предусмотренной приложением N 1 к Правилам
|
||
|
письмо производителя, содержащее мотивированное обоснование необходимости изменения в составе комплектующих, с указанием нового перечня комплектующих, подтверждающих отсутствие влияния на функциональные характеристики медицинского изделия
|
||
|
проекты инструкций по применению (руководство пользователя) медицинского изделия на государственном языке государства - члена Евразийского экономического союза и русском языке
|
||
|
обновленная спецификация с указанием перечня комплектующих и расходных материалов по утвержденной форме
|
||
|
опись документов
|
||
|
в случае добавления комплектующего, являющегося медицинским изделием, - образцы такого комплектующего (в случае стерильного комплектующего предоставляется весь комплект таких образцов) и нормативная документация на него
|
||
|
4. Показания по применению, области применения, противопоказания, побочные эффекты
|
безопасность применения медицинского изделия должна сохраняться и подтверждаться данными исследований, клинической безопасности и качества
|
заявление о внесении изменений по форме, предусмотренной приложением N 7 к Правилам
|
|
копия регистрационного удостоверения, оформленного по единой форме, предусмотренной приложением N 1 к Правилам
|
||
|
письмо производителя, содержащее мотивированное обоснование необходимости изменения показаний по применению медицинского изделия
|
||
|
проекты инструкций по применению (руководство пользователя) медицинского изделия
|
||
|
ранее утвержденная инструкция по применению (руководство пользователя) медицинского изделия
|
||
|
полноцветные макеты упаковок, этикеток, стикеров (при необходимости) (на электронном носителе CD в формате JPEG)
|
||
|
результаты клинических (медицинских) испытаний, отражающие внесенные изменения
|
||
|
опись документов
|
||
|
5. Сведения о производителе медицинского изделия
|
отсутствуют изменения в производственном процессе или спецификациях, включая методы испытания
|
заявление о внесении изменений по форме, предусмотренной приложением N 7 к Правилам
|
|
документ <*>, удостоверяющий регистрацию медицинского изделия в стране производителя (регистрационное удостоверение, сертификат свободной продажи, сертификат на экспорт и др.) с внесенными изменениями
|
||
|
документ <*>, подтверждающий внесение изменений (с указанием даты внесения изменений)
|
||
|
документ <*>, подтверждающий соответствие условий производства национальным и/или международным стандартам (GMP, ISO EN)
|
||
|
документ <*>, подтверждающий соответствие медицинского изделия национальным или международным стандартам, класс потенциального риска (декларация соответствия; сертификат соответствия)
|
||
|
копия регистрационного удостоверения по форме, предусмотренной приложением N 1 к Правилам
|
||
|
письмо производителя, удостоверяющее, что производственный процесс и контроль за качеством и безопасностью готового продукта остаются без изменений, с указанием даты внесения изменений
|
||
|
проекты инструкций по применению изделий (руководство пользователя) медицинского изделия
|
||
|
макет маркировки
|
||
|
опись документов
|
||
|
6. Спецификация производителя или технические условия (при наличии), которым соответствует медицинское изделие, и (или) эксплуатационная документация медицинского изделия
|
отсутствуют изменения в производственном процессе или спецификациях, включая методы испытания
|
заявление на внесение изменений по форме, предусмотренной приложением N 7 к Правилам
|
|
копия регистрационного удостоверения по форме, предусмотренной приложением N 1 к Правилам
|
||
|
письмо-обоснование производителя о вносимых изменениях
|
||
|
данные по стабильности (для медицинского изделия) не менее чем на 3 сериях (отчет, обосновывающий срок годности медицинского изделия) (при необходимости)
|
||
|
проект инструкции по применению (руководства пользователя) медицинского изделия (при необходимости)
|
||
|
полноцветные макеты упаковок, этикеток, стикеров (при необходимости)
|
||
|
нормативная документация <*> с внесенными изменениями, регламентирующая качество конечного продукта, сертификат анализа и методики контроля конечного продукта (при необходимости)
|
||
|
протокол технических испытаний или испытаний (исследований) с целью оценки биологического действия с учетом изменений, внесенных в нормативную документацию (при необходимости)
|
||
|
опись документов
|
--------------------------------
<*> Документы представляются с обязательным аутентичным переводом на русский язык, заверенным нотариально.
Приложение N 9
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ЭКСПЕРТНОЕ ЗАКЛЮЧЕНИЕ
о возможности (невозможности) внесения изменений
в регистрационное досье на медицинское изделие
1. Наименование медицинского изделия.
2. Производитель, страна.
3. Производственная площадка, страна.
4. Область применения и назначение.
5. Вид медицинского изделия в соответствии с номенклатурой медицинских изделий, применяемой в Евразийском экономическом союзе.
6. Класс потенциального риска применения медицинского изделия.
7. Номер регистрационного удостоверения.
8. Дата выдачи регистрационного удостоверения.
9. Вносимые изменения.
|
Тип вносимого изменения
|
Данные, внесенные в досье при регистрации
|
Вносимые изменения
|
Обоснование заявителя для внесения изменений
|
|
1. Изменение наименования производителя, места (мест) производства для части и/или всего производственного процесса
|
|||
|
2. Изменение наименования медицинского изделия
|
|||
|
3. Изменение состава принадлежностей, комплектующих и (или) расходных материалов
|
|||
|
4. Изменение показаний по применению, области применения, противопоказаний; побочных эффектов
|
|||
|
5. Изменение сведений о производителе медицинского изделия
|
|||
|
6. Изменение в спецификации производителя или технических условий (при наличии), которым соответствует медицинское изделие, и (или) эксплуатационной документации медицинского изделия
|
10. Анализ и оценка данных, обосновывающих внесение изменений.
11. Анализ рисков при внесении изменений (заключение о возможных рисках при внесении изменений).
12. Заключение о рекомендации или отказе в рекомендации к регистрации вносимых изменений.
13. Ф. И. О., должность, ученая степень (звание) (при наличии), подпись экспертов, проводивших экспертизу.
14. Дата составления отчета.
15. Подпись руководителя экспертной организации.
16. Печать экспертной организации.
Приложение N 10
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАЯВЛЕНИЯ
ОБ ОТМЕНЕ ДЕЙСТВИЯ (АННУЛИРОВАНИИ) РЕГИСТРАЦИОННОГО
УДОСТОВЕРЕНИЯ МЕДИЦИНСКОГО ИЗДЕЛИЯ
(на бланке организации)
В уполномоченный орган
референтного государства
ЗАЯВЛЕНИЕ
об отмене действия (аннулировании) регистрационного удостоверения
медицинского изделия
___________________________________________________________________________
(полное и сокращенное (при наличии) наименования заявителя, в том числе
фирменное наименование, организационно-правовая форма юридического лица)
Просит отменить действие регистрационного удостоверения (аннулировать
регистрационное удостоверение) медицинского изделия
___________________________________________________________________________
(наименование медицинского изделия (с указанием принадлежностей,
необходимых для применения медицинского изделия по назначению))
___________________________________________________________________________
(дата регистрации медицинского изделия и номер регистрационного
удостоверения)
в связи с _________________________________________________________________
(указывается причина)
___________________________________________________________________________
(Ф. И. О. руководителя производителя медицинского изделия
(его уполномоченного представителя))
"__" _____________ 20__ г. М.П. _____________________
(подпись)
Приложение N 11
к Правилам регистрации и экспертизы
безопасности, качества и эффективности
медицинских изделий
ФОРМА ЗАЯВЛЕНИЯ
О ВЫДАЧЕ ДУБЛИКАТА РЕГИСТРАЦИОННОГО УДОСТОВЕРЕНИЯ
НА МЕДИЦИНСКОЕ ИЗДЕЛИЕ
(на бланке организации)
В уполномоченный орган
референтного государства
ЗАЯВЛЕНИЕ
о выдаче дубликата регистрационного удостоверения
на медицинское изделие
|
1. Наименование медицинского изделия
(с указанием принадлежностей, необходимых для применения медицинского изделия по назначению, - в виде приложения к заявлению, заверенного печатью и подписью руководителя)
|
|
|
I. В отношении производителя медицинского изделия
|
|
|
2. Организационно-правовая форма и полное наименование юридического лица
|
|
|
3. Сокращенное наименование юридического лица
(при наличии)
|
|
|
4. Фирменное наименование юридического лица
(при наличии)
|
|
|
5. Место нахождения (адрес) юридического лица
|
|
|
6. Номер телефона
|
|
|
7. Адрес электронной почты юридического лица
(при наличии)
|
|
|
8. Идентификационный номер налогоплательщика
|
|
|
II. В отношении уполномоченного представителя производителя
|
|
|
9. Организационно-правовая форма и полное наименование юридического лица
|
|
|
10. Сокращенное наименование юридического лица
(при наличии)
|
|
|
11. Фирменное наименование юридического лица
(при наличии)
|
|
|
12. Место нахождения (адрес) юридического лица
|
|
|
13. Номер телефона
|
|
|
14. Адрес электронной почты юридического лица
(при наличии)
|
|
|
15. Идентификационный номер налогоплательщика
|
|
|
16. Место производства медицинского изделия
|
|
|
17. Назначение медицинского изделия, установленное производителем
|
|
|
18. Вид медицинского изделия в соответствии с номенклатурой медицинских изделий, применяемой в Евразийском экономическом союзе
|
|
|
19. Класс медицинского изделия в соответствии с правилами классификации медицинских изделий в зависимости от потенциального риска применения
|
|
|
20. Способ получения информации, связанной с процедурой регистрации медицинского изделия
|
|
|
21. Способ получения регистрационного удостоверения медицинского изделия
|
на бумажном носителе лично
|
|
на бумажном носителе направить заказным почтовым отправлением с уведомлением о вручении
|
|
|
в форме электронного документа
|
|
|
иное
|
|
|
22. Причина выдачи дубликата
|
|
|
23. Сведения об уплате государственной пошлины
(указывается по инициативе заявителя):
|
|
|
дата и номер платежного поручения
|
|
___________________________________________________________________________
(Ф. И. О. руководителя производителя медицинского изделия
(его уполномоченного представителя))
"__" _____________ 20__ г. М.П. _____________________
(подпись)