СОВЕТ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 3 ноября 2016 г. N 89
ОБ УТВЕРЖДЕНИИ ПРАВИЛ
ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ БИОЛОГИЧЕСКИХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА
В соответствии со статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, пунктом 88 приложения N 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 98, и Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 108 "О реализации Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза" Совет Евразийской экономической комиссии решил:
1. Утвердить прилагаемые Правила проведения исследований биологических лекарственных средств Евразийского экономического союза.
2. Настоящее Решение вступает в силу по истечении 10 календарных дней с даты вступления в силу Протокола, подписанного 2 декабря 2015 года, о присоединении Республики Армения к Соглашению о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, но не ранее чем по истечении 10 календарных дней с даты официального опубликования настоящего Решения.
Члены Совета Евразийской экономической комиссии:
|
От Республики Армения
|
От Республики Беларусь
|
От Республики Казахстан
|
От Кыргызской Республики
|
От Российской Федерации
|
|
В.ГАБРИЕЛЯН
|
В.МАТЮШЕВСКИЙ
|
А.МАМИН
|
О.ПАНКРАТОВ
|
И.ШУВАЛОВ
|
Утверждены
Решением Совета
Евразийской экономической комиссии
от 3 ноября 2016 г. N 89
ПРАВИЛА
ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ БИОЛОГИЧЕСКИХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА
I. Введение
Настоящие Правила разработаны на основе актов, входящих в право Евразийского экономического союза (далее - Союз), и международных рекомендаций (Всемирной организации здравоохранения, Международной конференции по гармонизации технических требований при регистрации лекарственных препаратов по медицинскому применению, Европейского агентства по лекарственным средствам) в целях создания и поддержания системы взаимного признания государствами - членами Союза (далее - государства-члены) результатов фармацевтических и биологических испытаний, доклинических и клинических исследований лекарственных препаратов для их дальнейшей регистрации. Основополагающим принципом проведения исследований биологических лекарственных препаратов является выполнение требований международных договоров и актов, составляющих право Союза, или законодательства государств-членов в данной области (при отсутствии нормативно-правового регулирования вопросов в праве Союза).
Выбор вида и объема фармацевтических и биологических испытаний, доклинической и клинической оценки новых лекарственных препаратов должен основываться на актуальной научной информации.
Основной целью настоящих Правил является поддержание высокого уровня проведения фармацевтических и биологических испытаний, доклинических и клинических исследований, их единообразия. Настоящие Правила предназначены для упрощения сбора и представления данных, прилагаемых к заявлениям о регистрации биологических лекарственных препаратов в государствах-членах. Настоящие Правила следует применять одновременно с международными договорами и актами в сфере обращения лекарственных средств, составляющими право Союза (в том числе с правилами надлежащих фармацевтических практик Союза).
Требования настоящих Правил распространяются на разработчиков, исследователей и производителей лекарственных средств, а также держателей регистрационных удостоверений лекарственных препаратов и их доверенных лиц, уполномоченные органы в сфере обращения лекарственных средств и экспертные организации государств-членов.
Проведение фармацевтических и биологических, доклинических и клинических исследований биологических лекарственных препаратов должно проводиться с учетом особенностей, связанных как со свойствами и природой этих препаратов (инактивированные и живые вакцины, биотехнологические лекарственные препараты на основе рекомбинантных белков, аллергены, препараты из плазмы человека, гетерологичные сыворотки и др.), так и со сферой их применения в клинической практике.
Классификация биологических лекарственных препаратов
К биологическим лекарственным препаратам относятся иммунологические (иммунобиологические) лекарственные препараты, биотехнологические лекарственные препараты, лекарственные препараты, полученные из плазмы человека, препараты пробиотиков (эубиотиков), препараты бактериофагов, высокотехнологические лекарственные препараты, а также лекарственные препараты, содержащие следующие активные фармацевтические субстанции нерекомбинантного происхождения, произведенные или выделенные из биологических источников (тканей, жидкостей и органов человека, сырья животного происхождения, микроорганизмов или продуктов их жизнедеятельности): хорионический гонадотропин человека, менотропин, урофоллитропин, стрептокиназа, стрептодорназа, урокиназа, апротинин, гиалуронидаза, протамин, ботулинические токсины, БЦЖ для внутрипузырной инстилляции, гепарин, хондроитина сульфат, далтепарин, эноксапарин, надропарин, тинзапарин, ревипарин, парнапарин, цертопарин, данапароид натрия, панкреатин, аспарагиназа, анти-T-лимфоцитарный иммуноглобулин для медицинского применения животного происхождения, поликлональный овечий антидигоксин, интерферон альфа человека, бычий инсулин, свиной инсулин, пепсин, трипсин, химотрипсин, глюкагон животного происхождения, лизаты бактерий, низкомолекулярные фракции крови телят, церебролизин, фосфолипиды животного происхождения, коллагеназа, дезоксирибонуклеаза животного происхождения, фибринолизин животного происхождения, компоненты клеток микроорганизмов.
Приведенный перечень не является закрытым, в соответствующих случаях к биологическим лекарственным препаратам могут быть отнесены лекарственные препараты, содержащие фармацевтические субстанции, не указанные выше.
Настоящие Правила не распространяются на цельную кровь, плазму и клетки крови человека (за исключением плазмы крови, приготовленной по методу, включающему промышленный процесс), а также на антибиотики, если в других актах, входящих в право Союза, не указаны ссылки на настоящие Правила.
II. Определения
Для целей настоящих Правил используются понятия, которые означают следующее:
"аллерген" - молекула, способная индуцировать IgE-ответ и (или) аллергическую реакцию I типа;
"банк клеток" - набор контейнеров с однородным по составу содержимым, который хранят в определенных условиях. Каждый контейнер содержит аликвоту одного пула клеток;
"биоаналогичный лекарственный препарат", "биоаналог", "биоподобный лекарственный препарат", "биосимиляр" - биологический лекарственный препарат, который содержит версию действующего вещества зарегистрированного биологического оригинального (референтного) препарата и для которого продемонстрировано сходство (подобие) на основе сравнительных исследований с оригинальным (референтным) препаратом по показателям качества, биологической активности, эффективности и безопасности;
"биологические лекарственные препараты" - лекарственные препараты, действующее вещество которых произведено или выделено из биологического источника и для характеристики свойств и контроля качества которых необходимо сочетание биологических и физико-химических методов анализа с оценкой производственного процесса и методов его контроля;
"биотехнологический лекарственный препарат" - лекарственный препарат, произведенный при помощи биотехнологических процессов и применения методов с использованием технологии рекомбинантной ДНК, контролируемой экспрессии генов, кодирующих выработку биологически активных белков, гибридомных технологий, моноклональных антител или других биотехнологических процессов;
"вакцина" - лекарственный препарат, предназначенный для формирования активного иммунитета с целью профилактики инфекционных заболеваний у человека;
"вирус" - внутриклеточно реплицирующиеся инфекционные агенты, которые потенциально патогенны, обладают только одним видом нуклеиновой кислоты (либо РНК, либо ДНК), не способны расти и не подвергаются бинарному делению, размножаются в форме своего генетического материала;
"вирусоподобные частицы" - структуры, различимые под электронным микроскопом, морфология которых сходна с морфологией известных вирусов;
"главный банк клеток", "ГБК" - аликвота из единого пула клеток, которая, как правило, получается из выбранного клона клеток в заданных условиях, распределяется по множеству контейнеров и хранится в заданных условиях. ГБК применяют для получения всех рабочих банков клеток. При отсутствии соответствующего обоснования испытания, проводимые на новом ГБК (полученном из предшествующего первоначального клона клеток или ГБК), должны совпадать с результатами испытаний предшествующего ГБК;
"иммуногенность" - способность лекарственного препарата (как правило, биологического) вызывать иммунный ответ на себя и родственные белки или приводить к иммунологически опосредованным нежелательным клиническим явлениям;
"иммуногенность вакцины" - фармакологический эффект вакцины, заключающийся в выработке иммунного ответа на определенный возбудитель инфекционного заболевания или продукт его жизнедеятельности и определяющий профилактическую эффективность вакцины;
"иммунологический лекарственный препарат", "иммунобиологический лекарственный препарат" - лекарственный препарат, предназначенный для формирования активного или пассивного иммунитета, или диагностики наличия иммунитета, или диагностики (выработки) специфического приобретенного изменения иммунологического ответа на аллергизирующие вещества. К иммунологическим (иммунобиологическим) лекарственным препаратам относятся вакцины, анатоксины, токсины, сыворотки, иммуноглобулины и аллергены;
"инактивация" - снижение инфицирующей способности вируса, обусловленное его химической или физической модификацией;
"исследование процесса очистки от вирусов" - исследование очистки от вирусов, в котором используют неспецифичные, "релевантные" и (или) специфичные "модельные" вирусы для определения способности процесса производства элиминировать и (или) инактивировать вирусы;
"исследование сопоставимости" - исследование, направленное на подтверждение отсутствия клинически значимых различий между биологическим лекарственным препаратом после внесения изменений в технологию (процесс) его производства относительно биологического лекарственного препарата, произведенного по неизмененной технологии;
"исследование сопоставимости в рамках оценки биоподобия" (biosimilarity exercise) - комплекс фармацевтических и биологических испытаний, доклинических и клинических исследований, направленных на подтверждение отсутствия клинически значимых различий между оригинальным (референтным) и биоаналогичным (биоподобным) лекарственными препаратами в процессе разработки последнего;
"клетки-продуценты" - клетки, используемые для производства препарата;
"клеточная линия" - тип клеточной популяции, образующейся путем последовательного субкультивирования популяции первичных клеток, из которой можно сформировать банк клеток;
"клеточный возраст in vitro", "продолжительность жизни клеток in vitro" - период с момента оттаивания контейнера с ГБК до получения производственной емкости, измеряемый истекшим хронологическим временем, степенью удвоения популяции клеток или числом пассажей клеток при субкультивировании с помощью определенной процедуры разведения культуры;
"клеточный субстрат" - клетки, используемые для производства продукта;
"лекарственные препараты, полученные из плазмы" - лекарственные препараты, произведенные промышленным способом из компонентов крови человека. К данным препаратам относятся альбумины, факторы свертывания крови и иммуноглобулины человеческого происхождения;
"линия диплоидных клеток", "диплоидная клеточная линия" - клеточная линия, которая обладает конечной продолжительностью жизни in vitro, и в которой хромосомы являются парными (эуплоидными) и структурно идентичными хромосомам видов, из которых эти клетки были получены;
"неспецифичный "модельный" вирус" - вирус, который используется для установления характеристик процесса очистки от вирусов, целью которого является характеристика общей способности процесса производства элиминировать и (или) инактивировать вирусы, то есть характеристика устойчивости (надежности) процесса очистки;
"неэндогенный вирус" - вирус, попавший в ГБК из внешних источников;
"опытно-промышленная серия", "пилотная серия", "валидационная серия" - серия активной фармацевтической субстанции или лекарственного препарата, изготовленная способом, который полностью соответствует и воспроизводит промышленный способ производства;
"очистка от вирусов" - устранение вируса путем физической элиминации частиц вируса или инактивация его инфицирующей способности;
"перевиваемая клеточная линия" - клеточная линия, обладающая бесконечной возможностью роста. Такую клеточную линию часто называют бессмертной;
"посторонний вирус" - непреднамеренно привнесенный контаминирующий вирус;
"предельный для производства клеточный возраст in vitro" - величина, равная или не превышающая предлагаемую продолжительность культивирования клеток при указанных для производственного процесса условиях;
"препараты аллергенов" - лекарственные препараты, содержащие аллергены или производные аллергенов, применяющиеся с целью диагностики in vivo или лечения аллергических заболеваний;
"препараты бактерофагов" - препараты на основе вирусов, способных проникать в бактериальную клетку, размножиться в ней, вызывать ее лизис или переход в состояние лизогении (фагоносительства);
"промышленная серия" - серия фармацевтической субстанции или лекарственного препарата, произведенная промышленным способом с использованием производственного оборудования на производственной площадке, указанной в регистрационном досье лекарственного препарата;
"рабочий банк клеток", "РБК" - банк клеток, который состоит из аликвот гомогенной суспензии клеток, полученных при культивировании ГБК в заданных условиях;
"релевантный" вирус" - вирус, который используется для исследований анализа процесса и который либо является выявленным вирусом, либо принадлежит к виду вирусов, которые контаминировали или с высокой вероятностью могут контаминировать клеточный субстрат или какие-либо реактивы или материалы, использованные в процессе производства;
"родительские клетки", "исходные клетки", "клетки-хозяина" (host-cells) - клетки, используемые для создания клеточного субстрата или промежуточной клеточной линии. В случае использования экспрессионных систем на основе микроорганизмов родительские клетки обычно называются клетками хозяина. В отношении гибридом родительская клетка также называются сливающимися клетками;
"специфичный "модельный" вирус" - вирус, являющийся близкородственным выявленному или подозреваемому вирусу (принадлежит тому же роду или семейству), обладающий схожими с ним физическими и химическими свойствами;
"элиминация вирусов" - физическое отделение вирусных частиц от целевого продукта;
"эндогенный вирус" - вирус, геном которого является частью генеративной линии вида, являющегося источником клеточной линии, и который ковалентно интегрирован в геном животного, из которого получена родительская клеточная линия. В целях настоящих Правил к этой категории относятся преднамеренно введенные неинтегрированные вирусы, например, вирус Эпштейна-Барр, используемый для иммортализации клеточного субстрата, или папилломавирус коров.
В настоящих Правилах используются следующие сокращения:
|
ВИЧ
|
- вирус иммунодефицита человека
|
|
ВОЗ
|
- Всемирная организация здравоохранения
|
|
ВЭЖХ
|
- высокоэффективная жидкостная хроматография
|
|
ГБК
|
- главный банк клеток
|
|
ГЗТ
|
- гиперчувствительность замедленного типа
|
|
ГНТ
|
- гиперчувствительность немедленного типа
|
|
ДИ
|
- доверительный интервал
|
|
ДНК
|
- дезоксирибонуклеиновая кислота
|
|
ИФА
|
- иммуноферментный анализ
|
|
ЛД50
|
- доза, вызывающая гибель 50% животных
|
|
МЕ
|
- международная единица
|
|
ПУР
|
- план управления рисками
|
|
ПЦР
|
- полимеразная цепная реакция
|
|
РНК
|
- рибонуклеиновая кислота
|
|
ТГЭ
|
- трансмиссивная губчатая энцефалопатия
|
|
ФД
|
- фармакодинамика
|
|
ФК
|
- фармакокинетика
|
|
ЦНС
|
- центральная нервная система
|
|
ЦПКП
|
- целевой профиль качества лекарственного препарата
|
|
HRQoL
|
- связанное со здоровьем качество жизни
|
|
Ig
|
- иммуноглобулин
|
|
IL
|
- интерлейкин
|
|
TGF
|
- трансформирующий фактор роста бета
|
|
VAS
|
- визуальная аналоговая шкала
|
|
Th
|
- лимфоциты T-хелперы
|
III. Основной текст правил
Глава 1. Выделение и характеристика клеточных субстратов,
используемых при производстве биотехнологических
(биологических) препаратов
1. Введение, область применения
Ряд проблем, связанных с качеством получаемых из клеток биологических лекарственных препаратов, обусловлены наличием посторонних контаминантов или свойствами клеток, используемых в производстве препарата. Для препаратов, получаемых с применением технологии рекомбинантной ДНК, кроме того, характерны проблемы качества, связанные с экспрессирующей конструкцией клеточного субстрата. Таким образом, свойства клеточного субстрата и процессов, затрагивающих клеточный субстрат, в результате могут влиять на качество лекарственного препарата и безопасность его применения. Кроме того, эффективный контроль качества этих препаратов требует надлежащей проверки всех манипуляций с клеточным субстратом.
Настоящая глава дополняет другие главы настоящих Правил и рекомендации Евразийской экономической комиссии (далее - Комиссия), что позволяет обеспечить всесторонний подход к оценке результатов качества, связанных с биологическими параметрами технологии получения препаратов из культур клеток Metazoa и микроорганизмов.
Действие настоящей главы распространяется на клеточные субстраты, в отношении которых создана система банков клеток. Для целей настоящих Правил под клеточным субстратом понимаются микробные клетки или линии клеток, выделенные из источников человеческого или животного происхождения, обладающие полным потенциалом, необходимым для продуцирования в условиях in vivo или ex vivo биотехнологических (биологических) препаратов для медицинского применения. Реактивы для диагностического применения в условиях in vitro в настоящей главе не рассматриваются. Источники клеточных линий животного происхождения включают в себя все организмы, принадлежащие к подцарству Metazoa. Описаны перевиваемые клеточные линии с неограниченной продолжительностью жизни in vitro, диплоидные клетки с ограниченным сроком жизни in vitro, а также клеточные субстраты микробиологического происхождения (бактерии, грибы, дрожжи и другие одноклеточные организмы).
Настоящая глава включает в себя общие вопросы по стандартам получения клеточных линий человека и животных, микробных клеток, а также вопросы формирования и характеристики банков клеток, используемых для производства биологических препаратов, рекомендации по получению данных, которые необходимо включить в регистрационное досье при подаче заявления на регистрацию биологического препарата в государствах-членах.
Настоящая глава касается биологических препаратов, полученных из клеток, культивируемых из банков клеток, за исключением таких микробных метаболитов, как антибиотики, аминокислоты, углеводы и прочие низкомолекулярные вещества. Банки клеток, используемые для получения препаратов для генной терапии или вакцин, должны соответствовать требованиям, указанным в настоящей главе. Некоторые биологические препараты (например, определенные вирусные вакцины) производят на первичных клеточных культурах, полученных непосредственно из тканей или органов животных. Первичные клетки не используются для создания банка клеток, и поэтому настоящие Правила на них не распространяется. Однако в приложении приводятся другие подходы, которые могут быть применимы к таким первичным клеткам.
2. Требования к клеточным субстратам
2.1. Источник, история и получение клеточного субстрата
2.1.1. Необходимо представить первичную документацию, в которой описывается общая информация о клеточном субстрате, используемом для производства биологического препарата, а также о каждой родительской клеточной линии, из которой клеточный субстрат был выделен полностью или частично. Мероприятия, проводимые на этапе научных исследований и разработок клеточного субстрата, могут оказывать существенное влияние на риски, связанные с использованием в производстве конкретного клеточного субстрата. Представленная в связи с этим информация облегчает всестороннюю оценку рисков, позволяющую гарантировать качество и безопасность применения препарата.
Все производимые с клеточным субстратом манипуляции необходимо подробно документировать на протяжении всего процесса разработки. Описание истории клетки является одним из множества инструментов, используемых при установлении характеристики клеточного субстрата. Как правило, недостаточность данных об истории клеток не может служить препятствием для регистрации лекарственного препарата, но отсутствие достаточного объема данных может в результате привести к повышенной зависимости от других методов, используемых для характеристики клеточного субстрата.
2.1.2. Происхождение, источник и история клеток.
Необходимо указать источник клеток (лабораторная коллекция или коллекция культур), из которого был получен клеточный субстрат, и привести соответствующие ссылки на научный литературный источник. Предпочтительны данные, полученные непосредственно из исходной лаборатории. Если эта информация недоступна, можно использовать литературные данные.
Для клеточных линий человека необходимо описать следующие характеристики первоначального донора: происхождение ткани или органа, этническое и географическое происхождение, возраст, пол и общее физиологическое состояние. При наличии следует привести данные о состоянии здоровья или анамнез донора наряду с результатами любого тестирования донора на наличие патогенных агентов. Поскольку для диплоидных фибробластов человека возраст донора может влиять на продолжительность жизни клеточной линии in vitro, эти данные (при их наличии) следует представлять. При использовании линии животных клеток в описании источника необходимо указать виды, породы, условия разведения, происхождение ткани или органа, географическое происхождение, возраст, пол, а также результаты тестирования на наличие патогенных агентов и общее физиологическое состояние первичного донора.
При использовании микроорганизмов производители должны указать их вид, штамм и известные генотипические и фенотипические характеристики организма, из которого был выделен клеточный субстрат. Производители также должны привести данные по патогенности, токсигенности и прочую информацию о биологической опасности (если она имеет место).
Должна быть документально оформлена история культивирования клеток. Наряду с процедурами культивирования клеток in vitro и всеми процедурами создания клеточных линий (например, использование физических, химических, биологических методов или введение нуклеотидных последовательностей) необходимо описать метод, первоначально использованный для выделения клеток. Необходимо описать все имевшие место генетические манипуляции или генетический отбор (селекцию). Вся доступная информация, относящаяся к идентификации, характеристикам этих клеток и результатам их испытания на наличие эндогенных или посторонних агентов, тоже должна быть предоставлена.
Для перевиваемых клеточных линий, полученных от Metazoa, обычно бывает достаточно определить продолжительность культивирования путем оценки либо числа удвоений популяции, либо числа субкультивирований при определенном коэффициенте разведения или времени культивирования (в днях). Для линий диплоидных клеток, обладающих ограниченным сроком жизни in vitro, важна точная оценка числа удвоений на протяжении всех стадий исследования, разработки и производства. Для клеток микроорганизмов считается достаточным представление документации, содержащей сведения по определению частоты субкультивирования после создания клеточного субстрата.
В отношении получения клеточного субстрата заявители должны провести тщательный анализ процессов, при использовании которых возможен занос инфекционных агентов. Должно быть представлено описание компонентов культуральной среды, особенно информация, касающаяся воздействия на клетки таких компонентов человеческого и животного происхождения, как сыворотка, ферменты, гидролизаты или другие живые клетки. Описание должно включать источник, метод приготовления и контроля, результаты испытаний и обеспечение качества. Могут быть приведены соответствующие ссылки на литературные источники, если таковые доступны. Эта информация позволит провести детальный анализ возможных путей проникновения посторонних агентов из указанных источников и станет составной частью анализа соотношения "польза - риск" для препарата.
2.1.3. Создание клеточного субстрата (получение клеток-продуцентов).
Ключевой стадией является выбор подходящей родительской клеточной линии. Для рекомбинантных препаратов родительской клеточной линией служит, как правило, клеточная линия - реципиент, не подвергшаяся трансфекции. В этой ситуации рекомендуется использование охарактеризованных банков родительских (исходных) клеток. Охарактеризованный банк родительских (исходных) клеток может представить данные, на основании которых может быть проведена оценка качества ГБК, особенно в случаях, когда множество клеточных субстратов образовано из одного и того же типа родительских клеток. Например, клеточная линия миеломы может быть заготовлена в качестве родительской (исходной) клеточной линии для гибридомы.
При окончательной разработке требуемых характеристик в процессе создания клеточного субстрата возможно использование одной или нескольких специфических процедур. К ним относятся, например, слияние (гибридизация) клеток, трансфекция, отбор клонов, выделение колоний, клонирование, амплификация генов и адаптация к специфическим условиям культивирования или средам. Информация, касающаяся методологии, используемой при разработке клеточного субстрата, может помочь в обеспечении понимания истории клеточного субстрата. Некоторые клеточные субстраты, например, диплоидные фибробласты человека, могут не требовать интенсивной обработки или предварительного клонирования перед созданием банка клеток.
В рекомбинантных препаратах клеточным субстратом служат трансфецированные клетки, содержащие требуемые последовательности, которые были клонированы из единственной клетки предшественника. При создании клеточных субстратов с использованием технологии рекомбинантной ДНК необходимо учитывать рекомендации главы 5.2 настоящих Правил. Для нерекомбинантных препаратов (в том числе нерекомбинантных вакцин) клеточным субстратом служат клетки из линии родительских (исходных) клеток, отобранных для создания ГБК, без дальнейшей модификации. Для препаратов, получаемых из гибридом, клеточным субстратом служит гибридомная клеточная линия, полученная путем слияния родительской клеточной линии миеломы с другими родительскими клетками, например, с иммунными клетками селезенки.
2.2. Создание банка клеток
Одним из наиболее важных преимуществ использования серийно субкультивируемых клеток для производства биотехнологических (биологических) препаратов является возможность иметь охарактеризованный общий источник для каждой производственной серии, то есть иметь законсервированный банк клеток. Производители могут создавать свои собственные банки клеток или получать их из внешних источников. Производители обязаны обеспечивать качество каждого банка клеток и проводить с каждым из них необходимые испытания.
2.2.1. Система банков клеток.
Концепция двухуровневой структуры банка клеток, в которой ГБК используется для РБК, как правило, принимается в качестве наиболее практичного подхода, обеспечивающего получение клеточного субстрата для непрерывного производства препарата. Производители должны описать стратегию обеспечения непрерывного получения клеток из банка (банков), включая ожидаемую скорость расходования банка клеток при производстве, ожидаемые интервалы между созданием новых банков клеток и показатели, по которым аттестуются (характеризуются) банки клеток.
Как правило, сначала создается ГБК, обычно непосредственно из исходного клона или из предварительного банка клеток, полученного из исходного клона. Приготовление банка клеток из клонов не является обязательным для определенных типов клеток (например, диплоидных клеток, продолжительность жизни которых in vitro ограничена), или при наличии других технических факторов, делающих клонирование клеток непрактичным, или в случаях, когда неклонированная клеточная популяция уже достаточно гомогенна для предполагаемого применения.
Для создания РБК используются один или более контейнеров с клетками из ГБК. Именно РБК, как правило, используется непосредственно для нужд производственного процесса. По мере необходимости из ГБК создаются дополнительные РБК. Свежеприготовленный РБК должен быть соответствующим образом квалифицирован путем установления его характеристик и проведения испытаний.
ГБК и РБК могут отличаться друг от друга по ряду параметров, например, компонентами культуральной среды и условиями культивирования. Кроме того, условия культивирования, используемые при приготовлении ГБК и РБК, могут отличаться от используемых в процессе производства. Если изменения процесса культивирования клеток не оказывают неблагоприятного воздействия на качество препарата, повторное клонирование клеток или повторное создание ГБК или РБК не требуется. Необходимо убедиться, что охарактеризованный банк обеспечивает получение продукта постоянного качества.
Допускается использование одноуровневого банка, состоящего только из ГБК и не содержащего РБК, например в случае, если для производства препарата каждый год требуется относительно небольшое число контейнеров с клетками.
В некоторых микробных экспрессирующих системах для каждой новой серии контейнеров с клеточным субстратом проводится новая трансформация с использованием аликвот тщательно испытанных банков клеток хозяина и банков плазмид для каждой новой трансформации. Кроме того, проводится испытание каждого банка трансформированного клеточного субстрата. Такой банк трансформированного клеточного субстрата рассматривается как ГБК и используется в качестве источника клеточного субстрата для производственного процесса. Банки клетки хозяина, банки плазмид и ГБК хранятся с помощью соответствующих методов консервации. Эта альтернативная система считается достаточной, поскольку трансформация бактерий и дрожжей, как правило, представляет собой легко воспроизводимый процесс, в отличие от процедуры, применяемой для трансфекции клеток Metazoa. Производители должны предоставлять информацию о клетках хозяина, молекулах рекомбинантной ДНК (таких как плазмиды), методе трансформации и создании банка клеток, а также о результатах исследований по характеристике их свойств.
2.2.2. Процедуры создания и поддержания банка клеток.
Необходимо предотвратить использование контаминированного клеточного субстрата (или банка клеток) в процессе производства препарата и избежать снижения доступности или производства препарата, либо потерь времени при разработке, возникающих в результате необходимости повторного создания банка клеток, который оказался непригодным вследствие контаминации. Ни один режим испытаний банка клеток не позволяет выявлять все возможные контаминанты, поэтому использование описанных предупредительных мер во время создания банка клеток важно для обеспечения достаточной уверенности в отсутствии контаминации, а также для создания надежного источника клеточного субстрата.
Производители должны описать тип используемой системы создания банка, размер банка клеток, тип контейнера (флаконы, ампулы или другие подходящие емкости) и используемую систему укупорки, методы приготовления банка клеток, включая используемые криопротекторы и среду, а также условия криоконсервации и хранения.
Производители должны описать процедуры, используемые для предотвращения микробной контаминации и перекрестной контаминации другими типами клеток, присутствующими в лаборатории, а также описать процедуры, позволяющие отслеживать емкости банка клеток. К этим мероприятиям относится описание системы документации, а также системы маркировки, которая может выдержать процесс консервации, хранения и восстановления без потери информации, нанесенной на контейнер.
Производители должны описать технологию создания и поддержания банка клеток. Как правило, клетки готовятся для банка путем наращивания объемов культивирования за счет постепенного увеличения числа сосудов или использования сосудов большего размера до тех пор, пока не получится пул клеток, которого будет достаточно для создания необходимого числа контейнеров с клетками для банка клеток. Для того чтобы обеспечить однородный состав содержимого каждого контейнера, каждый пул клеток для создания банка должен быть приготовлен путем объединения клеток из всех сосудов для культивирования (при использовании более одного сосуда).
Аликвоты клеток, суспендированных в среде для консервации, переносятся из объединенного пула в стерильные контейнеры, которые затем укупориваются и хранятся в необходимых условиях. Например, клетки животных в средах, содержащих криопротектор, замораживаются в укупоренных контейнерах при заданных контролируемых условиях, а затем переносятся на хранение в жидком азоте, или его парах, или при соответствующих сверхнизких температурах. В зависимости от используемого организма допускается применять иные методы консервации и хранения. Однако они должны позволять поддерживать такую степень жизнеспособности клеток после разведения, которая была бы и постоянной и пригодной для производственных целей.
Для обеспечения постоянного, непрерывного производства лекарственных препаратов производители должны предусмотреть меры по защите от аварийных ситуаций, которые могут привести банк клеток в непригодное для использования состояние. К таким ситуациям относятся: пожары, перебои в электроснабжении и человеческий фактор. Производители должны описать планы предупредительных мероприятий для таких случаев. Например, к подобным мероприятиям можно отнести хранение контейнеров банка клеток в нескольких морозильных камерах, использование резервных источников питания, использование систем автоматизированного заполнения жидким азотом для контейнеров хранения, хранение части ГБК и РБК в удаленных помещениях или регенерация ГБК.
Исходной точкой для оценки возраста клеток in vitro в процессе производства должен служить момент оттаивания одного или более контейнеров с ГБК. В отношении линий диплоидных клеток продолжительность жизни клеток in vitro следует оценивать исходя из скорости удвоения клеточной популяции. Для диплоидных клеток следует определять допустимый уровень удвоения клеточной популяции, то есть уровень, при котором наступает биологическое старение.
2.3. Общие принципы установления характеристик
и испытания банков клеток
Установление характеристик и испытание клеточных субстратов из банков клеток представляет собой критическую составляющую контроля биологических и биотехнологических препаратов. Установление характеристик ГБК позволяет производителю оценить этот источник с точки зрения наличия клеток других клеточных линий, посторонних агентов, эндогенных агентов и молекулярных контаминантов (например, токсинов или антибиотиков из организма хозяина). Задача подобного тестирования состоит в подтверждении подлинности, чистоты и пригодности клеточного субстрата для использования в производстве. В некоторых случаях целесообразно проведение таких дополнительных испытаний, как проверка на туморогенность или кариотипирование. Программа проведения испытаний, выбранная для конкретного клеточного субстрата, может варьироваться в зависимости от биологических свойств клеток (например, потребности в питательных веществах для роста), истории культивирования клеточного субстрата (включая использование биологических реагентов человеческого или животного происхождения) и наличия подходящих аналитических методик. Объем исследований при установлении характеристик клеточного субстрата может влиять на вид или масштаб стандартных испытаний, необходимых на последующих стадиях производства. Для каждого ГБК производители должны однократно проводить испытания клеточного субстрата на подлинность и чистоту, а также испытание стабильности в ходе культивирования клеток для каждого подлежащего регистрации лекарственного препарата. Кроме того, испытания на чистоту и подлинность следует проводить однократно для каждого РБК. Заявители также должны принять во внимание положения главы 2 настоящих Правил. Необходимо провести соответствующие испытания из числа описанных ниже, их описание и полученные результаты необходимо представить в составе регистрационного досье.
При установлении характеристик клеточных линий, содержащих экзогенные экспрессирующие конструкции, созданные с использованием технологии рекомбинантной ДНК, следует руководствоваться также положениями главы 5.2 настоящих Правил. С помощью аналогичных методов целесообразно также провести анализ кодирующих последовательностей в некоторых клеточных линиях, при получении которых рекомбинантная ДНК не использовалась, если генные последовательности охарактеризованы и хорошо изучены. Однако не обязательно проводить исследования последовательностей, кодирующих такие сложные природные продукты, как, например, антигены микробных вакцин, моноклональные антитела из гибридом, семейства родственных генных препаратов.
При установлении характеристики клеточного субстрата и проведении испытаний производителям рекомендуется использовать современные методы и технологические достижения (при их доступности) при условии, что специфичность, чувствительность и прецизионность новых методов, по крайней мере, эквивалентна таковым параметрам существующих методов.
Производители могут проводить установление характеристик РБК вместо ГБК при условии представления соответствующего обоснования.
2.3.1. Испытания на подлинность.
Чтобы удостовериться в подлинности клеток в банке клеток, необходимо провести соответствующие испытания. При проверке подлинности могут быть оценены как фенотипические, так и генотипические характеристики, которые были установлены при разработке препарата. Не обязательно проводить все возможные испытания, однако объем выполненных испытаний должен быть обоснован. Испытания на подлинность обычно проводятся в отношении ГБК. Кроме того, ограниченный объем испытаний на подлинность, как правило, проводится в отношении каждого РБК.
2.3.1.1. Клетки Metazoa.
Морфологический анализ клеток человека или животных, выращиваемых в виде прикрепляющихся культур, может быть полезным инструментом в сочетании с другими тестами. В большинстве случаев для подтверждения происхождения клеточных линий человека или животных достаточно проведения изоферментного анализа, однако возможно проведение других тестов в зависимости от истории клеточной линии. Для верификации вида организма, из которого были получены клетки, могут быть использованы другие методики, включая, например, дифференциальное окрашивание хромосом (бэндинговая цитогенетика) или использование видоспецифичных антисывороток. Альтернативным подходом является демонстрация наличия уникальных маркеров, например, использование бэндинговой цитогенетики для обнаружения уникальной маркерной хромосомы или анализ ДНК для обнаружения геномного полиморфизма (например, полиморфизм длин рестрикционных фрагментов, вариабельность числа тандемных повторов или повторы геномных динуклеотидов). Достаточным испытанием на подлинность считается и установление вида животного, являющегося источником, и наличие изученных уникальных маркеров для клеточной линии. Экспрессия требуемого продукта может служить дополнением к подтверждению подлинности.
2.3.1.2. Клетки микроорганизмов.
Для большинства клеток микроорганизмов анализ роста на селективных средах обычно достаточен для подтверждения подлинности клетки-хозяина на уровне вида для банка клеток хозяина и банка трансформированных клеток. В случае E. coli, когда могут быть использованы разные штаммы, в качестве дополнительных методов испытания на подлинность следует рассматривать такой метод определения биологических характеристик, как фаготипирование. Для банков плазмид оценка подлинности может быть выполнена с помощью анализа экспрессирующей конструкции в соответствии с главой 5.2 настоящих Правил. Экспрессия требуемого продукта также может быть использована для подтверждения подлинности экспрессирующей конструкции.
2.3.2. Испытания на чистоту.
Важнейшим (критичным) аспектом разработки клеточной линии и создания банка клеток является оценка биологической чистоты ГБК и РБК, то есть доказательство того, что они свободны от посторонних микробных и клеточных контаминантов. При планировании и проведении этих испытаний необходимо учитывать влияние селективных агентов и антибиотиков на обнаружение посторонних микробных контаминантов.
2.3.2.1. Клетки Metazoa
Для проведения испытаний по оценке микробиологической чистоты (бионагрузки) (наличие бактерий и грибов) следует использовать индивидуальные контейнеры (1% от общего числа контейнеров, но не менее 2 контейнеров) для ГБК и РБК. В отношении остальных показателей следует использовать методологию испытаний микробиологических показателей или стерильности, предусмотренную Фармакопей Евразийского экономического союза (далее - Фармакопея Союза) или иных фармакопеях в соответствии с Концепцией гармонизации фармакопей государств - членов Евразийского экономического союза, утвержденной Решением Коллегии Евразийской экономической комиссии от 22 сентября 2015 года N 119.
ГБК и РБК должны быть испытаны на содержание микоплазм. Считаются достаточными методики Фармакопеи Союза, включающие в себя посев на агар и мясо-пептонный бульон, а также метод индикаторной клеточной культуры. Как правило, для проведения испытания достаточно клеток из одного контейнера. Для линий клеток немлекопитающих животных могут быть пригодны альтернативные методы контроля и (или) условия проведения испытаний. Для выбора соответствующей методики производители могут проконсультироваться с уполномоченными органами государств-членов.
Для обнаружения возможной контаминации вирусами должна быть разработана стратегия контроля клеточных субстратов на наличие вирусов, которая позволяет обнаруживать широкий спектр вирусов с использованием подходящих скрининг-тестов и соответствующих специфичных испытаний исходя из истории культивирования клеточной линии. Заявители должны применять требования главы 2 настоящих Правил. В отношении классов препаратов, не охваченных главой 2 настоящих Правил, следует руководствоваться рекомендациями ВОЗ по использованию животных клеток.
Чистота клеточных субстратов может быть нарушена в результате контаминации другими клеточными линиями, происходящими от того же или иного вида животных. Выбор необходимых испытаний зависит от существования возможности перекрестной контаминации другими клеточными линиями. В некоторых случаях необходимо поддержание роста разных клеточных линий в одной и той же лаборатории. При проведении процедур по созданию банка клеток, предусматривающих проведение открытых манипуляций, необходимо исключить одновременное проведение открытых манипуляций с другими клеточными линиями. Если в помещении, в котором осуществляется работа с банком клеток, находилась другая клеточная линия в момент, когда происходили открытые манипуляции с банком клеток (например, культивирование клеток, объединение, отбор аликвот выбранной клеточной линии), необходимо провести их испытания на наличие клеток (или продуктов, полученных из них) из второй клеточной линии. Как правило, указанные в подразделе 2.3.1 настоящей главы методы оценки подлинности клеток достаточны для выявления перекрестной контаминации другими клеточными линиями. Дополнительным подтверждением отсутствия перекрестной контаминации может быть получение препарата, соответствующего установленным требованиям.
2.3.2.2. Клетки микроорганизмов
При планировании и проведении специфических испытаний на посторонние микробные и клеточные контаминанты в банках клеток микроорганизмов необходимо учитывать свойства клеток в банках, возможные контаминанты, упоминаемые в научной литературе, источники, методы и материалы, используемые при культивировании клеток, а также другие организмы, находящиеся в лаборатории, в которой создается банк клеток. Например, возможно проведение визуальной оценки характеристик изолированных колоний с использованием разных микробиологических сред, поддерживающих и не поддерживающих рост клеточного субстрата. Тем не менее от производителей не требуется обязательно охарактеризовывать устойчивые мутанты клеточного субстрата, возникающие при таких исследованиях, или другие артефакты таких испытаний. Цель таких испытаний состоит, скорее, в выявлении существующих контаминантов.
2.3.3. Стабильность клеточного субстрата.
Одним из направлений изучения характеристик клеток является установление их пригодности для целевого использования в производстве. Существуют две проблемы, связанные со стабильностью клеточного субстрата: постоянство производства целевого продукта и сохранение производительности при его хранении в определенных условиях.
В целях оценки стабильности в ходе культивирования для производства необходимо провести исследования не менее чем в двух временных точках: первая - на клетках, подвергшихся минимальному количеству субкультивирований, вторая - на клетках при предельном для производства клеточном возрасте in vitro, описанном в регистрационном досье, или при превышении его. Предельный для производства клеточный возраст in vitro следует определять на основании данных, полученных на клетках-продуцентах, выращиваемых в опытно-промышленном или промышленном масштабе до предлагаемого предельного клеточного возраста in vitro или до возраста, превышающего его. Как правило, клетки-продуценты получают из РБК. Клетки из ГБК можно использовать при соответствующем обосновании. Оценка стабильности клеточного субстрата проводится обычно один раз для каждого регистрируемого лекарственного препарата.
Первостепенное значение имеет оценка способности клеточного субстрата обеспечить постоянство производства требуемого продукта. Вид проводимых испытаний и объекты исследований зависят от типа клеточного субстрата, продукта и методов культивирования. Для клеточных линий, содержащих экспрессирующие конструкции на основе рекомбинантной ДНК, неизменность кодирующей области экспрессирующей конструкции должна быть подтверждена на предназначенных для производства клетках, культивируемых до предельного клеточного возраста in vitro или дольше. Такая проверка проводится путем испытания нуклеотидной последовательности, для этих целей также могут быть использованы испытания продукта в соответствии с главой 5.2 настоящих Правил. Для нерекомбинантных клеточных линий, в которых кодирующая последовательность требуемого продукта уже была проанализирована на уровне ГБК или РБК, стабильность кодирующей белок последовательности на протяжении процесса производства должна быть подтверждена в клетках-продуцентах, культивированных до предполагаемого предельного клеточного возраста in vitro или дольше, путем либо испытания нуклеотидной последовательности, либо анализа очищенного белкового продукта.
Если продукт не может быть исследован указанным способом, для оценки стабильности клеточного субстрата можно использовать другие специфичные методы, например, установление морфологических характеристик, параметров роста, биохимических и иммунологических маркеров, прочих генотипических или фенотипических маркеров или выход требуемого продукта. В некоторых случаях, когда прямое сравнение характеристик ГБК с характеристиками клеток-продуцентов на предельном клеточном возрасте in vitro или при его превышении осуществить трудно или невозможно, для оценки стабильности клеточного субстрата на протяжении процесса производства можно сравнивать характеристики клеток на начальной стадии культивирования или производства с характеристиками клеток на предельном клеточном возрасте in vitro или при его превышении. Для подобного рода испытаний можно использовать такие показатели, как, например, скорость потребления кислорода или глюкозы, скорость выделения аммиака или лактата. Увеличение установленного предельного для производства клеточного возраста in vitro должно быть подтверждено данными для клеток, культивирование которых продолжалось до нового предложенного предельного клеточного возраста in vitro. Для линий диплоидных клеток должны быть представлены данные, устанавливающие конечный предел продолжительности жизни клеток из РБК в условиях, идентичных используемым при производстве.
Стабильность клеток в банке клеток в установленных условиях хранения обычно подтверждают во время производства материала для клинических исследований. Данные об определении жизнеспособности законсервированных клеток должны подтвердить, что клетки пережили процесс консервации и могут быть использованы для получения требуемого продукта. Данные о производстве материалов для клинических исследований позволяют удостовериться, что из восстановленных клеток можно получить требуемый продукт. Доступные данные должны быть отражены в документах регистрационного досье. Кроме этого, должен быть представлен план мониторинга стабильности банков клеток. Предлагаемый мониторинг допускается проводить, когда один или более контейнеров банков клеток, подвергшихся консервации, оттаивают в производственных целях при надлежащем наблюдении за постоянством свойств продукта или производства или когда один или более подвергшихся криоконсервации ГБК оттаивают с целью приготовления новых РБК (при этом новые РБК должным образом квалифицированы). Если производство не проводилось в течение длительного времени, определение жизнеспособности банка клеток, используемого в качестве источника продуцирующего субстрата, должно проводиться через определенный интервал времени, указанный в регистрационном досье. Если жизнеспособность клеточного субстрата снижается незначительно, как правило, дальнейшие испытания ГБК или РБК считаются нецелесообразными.
2.3.4. Кариотипирование и исследование туморогенности.
Для оценки безопасности линии диплоидных клеток или для характеристики новой клеточной линии проводятся кариотипирование и исследование туморогенных свойств в зависимости от типа клеток, свойств продукта и производственного процесса. Применение расширенного анализа для определения относительного содержания анеуплоидных клеток считается нецелесообразным. Нет необходимости проводить кариотипирование клеточных линий грызунов или новых клеточных линий, не относящихся к диплоидным. Как указано в подразделах 2.3.1 и 2.3.2 настоящей главы, цитогенетический анализ является достаточным методом для оценки подлинности клеточного субстрата или его чистоты. Повторное испытание туморогенности на клетках с уже подтвержденным туморогенным потенциалом проводить не требуется.
Кариотипирование и исследование туморогенности для высокоочищенных продуктов, не содержащих клетки, как правило, не требуются при условии, что доказано постоянство предельного остаточного содержания ДНК клетки-хозяина по результатам валидации процесса производства либо испытания серии при выпуске.
Как правило, проведение характеристики клеточного субстрата необходимо для препаратов, в которых нельзя исключить наличие живых клеток или которые подвергаются незначительной очистке в процессе выделения (например, некоторые живые вирусные вакцины). Целесообразность проведения исследований туморогенности и хромосомного анализа на новых клеточных субстратах для неочищенных препаратов следует оценивать в каждом конкретном случае. Возможность использования клеточных линий, обладающих известным туморогенным потенциалом или аномальным кариотипом, следует оценивать по соотношению "польза - риск" для каждого лекарственного препарата в случаях, когда он содержит клетки или когда степень его очистки невысока.
Если для производства препаратов используются генетически немодифицированные клетки MRC-5 или WI-38, нет необходимости характеризовать клеточные субстраты по кариологическим или туморогенным параметрам, поскольку эти клеточные линии достаточно охарактеризованы и результаты опубликованы. Тем не менее для каждого созданного РБК MRC-5 и WI-38 производители должны один раз подтвердить, что клетки, полученные при культивировании в условиях, аналогичных планируемому к использованию в производстве, являются диплоидными и имеют ожидаемую продолжительность жизни.
Для новых или ранее не охарактеризованных субстратов диплоидных клеток должно быть представлено подтверждение диплоидного кариотипа, а туморогенность должна быть установлена с использованием клеток из ГБК. Методы кариологического анализа и оценки туморогенности содержатся в документе ВОЗ "Требования к использованию клеток животных в качестве субстратов in vitro для производства биологических препаратов" (47-й отчет Экспертной комиссии ВОЗ по стандартизации в биологии, Женева, ВОЗ. Серия технических отчетов ВОЗ).
Приложение
к главе 1 Правил
проведения исследования
биологических лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ
К ПРЕДСТАВЛЕНИЮ ИНФОРМАЦИИ О ПЕРВИЧНЫХ КЛЕТОЧНЫХ КУЛЬТУРАХ
В РЕГИСТРАЦИОННОМ ДОСЬЕ ЛЕКАРСТВЕННОГО ПРЕПАРАТА
1. Введение
Изложенные в настоящем приложении принципы в большинстве случаев относятся к биотехнологическим (биологическим) препаратам, полученным из охарактеризованных банков клеток. Однако для производства ряда биологических лекарственных препаратов, в частности некоторых вирусных вакцин, используются первичные клеточные культуры.
Поскольку первичные клеточные культуры используются в рамках первого пассажа после создания из ткани источника, невозможно всесторонне охарактеризовать клетки до их использования, как это делается для клеточного субстрата, получаемого из банка клеток. Кроме того, биологические препараты, для производства которых используются первичные клеточные субстраты, зачастую не подвергаются интенсивной обработке (например, очистке). Несмотря на эти различия, для обеспечения пригодности и безопасности применения субстратов первичных клеток для создания биологических препаратов используется подход, во многих отношениях аналогичный изложенному в настоящих Правилах.
В настоящем приложении приводится информация, относящаяся к клеточному субстрату, которую следует включать в регистрационное досье лекарственного препарата, для производства которого используются первичные клетки. Эта информация разделяется на три основные категории:
информация, относящаяся к источнику ткани (органа) и другим исходным материалам животного происхождения, используемым для создания первичных клеточных субстратов;
информация, относящаяся к подготовке первичных клеточных субстратов;
информация, относящаяся к испытаниям, проводимым на первичных клеточных субстратах с целью обеспечения безопасности препарата.
2. Источники тканей и другие исходные материалы (сырье)
В регистрационном досье должна быть представлена информация о животных, используемых в качестве источника ткани для приготовления первичного клеточного субстрата. Ткань должна быть взята у здорового животного, прошедшего ветеринарный и лабораторный контроль для подтверждения отсутствия патогенных агентов. В случаях, когда это возможно, животные-доноры должны быть выращены в закрытых, беспатогенных (SPF) колониях или стадах. Животные, используемые в качестве доноров ткани, ранее не должны использоваться для экспериментальных исследований. Прежде чем использовать животных для получения клеток, они должны пройти обязательный карантинный контроль на протяжении установленного периода времени. Производители должны получить разъяснения уполномоченных органов государств-членов по вопросам, касающимся особых требований.
Необходимо представить информацию о материалах и компонентах, используемых для получения субстратов первичных клеток, включая указание типа и источника реагентов человеческого и животного происхождения. Необходимо включить описание испытаний, проведенных с компонентами животного происхождения, чтобы удостовериться в отсутствии поддающихся обнаружению контаминантов и посторонних агентов.
3. Приготовление первичных клеточных субстратов
Необходимо описать методы, используемые для выделения клеток из ткани, создания первичных культур клеток и их поддержания.
4. Испытания первичных клеточных субстратов
Необходимо описать испытания, которые проводятся на первичных клеточных субстратах для того, чтобы оценить возможность их использования в производстве. Поскольку природа первичных клеточных субстратов исключает возможность всесторонней проверки и определения характеристик перед их использованием, параллельно проводятся испытания с целью подтверждения отсутствия в этих субстратах посторонних агентов. Таким образом, испытания в целях подтверждения отсутствия посторонних агентов в таких субстратах проводятся параллельно и могут включать в себя наблюдение за производством или неинфицированными контрольными культурами до, в ходе и после осуществления процесса производства, инокуляцию культуральной жидкости после производства и из неинфицированных контрольных культур в различные восприимчивые индикаторные клеточные культуры, способные обнаруживать широкий круг вирусов с последующим изучением цитопатических изменений и испытаниями на гемадсорбирующие вирусы, а также прочие испытания на специфичные агенты (например, значимые ретровирусы) при необходимости. Дополнительные сведения о специфичных испытаниях на вирусы описаны в соответствующих национальных (региональных, международных) руководствах.
Надлежащие режимы и методы испытаний клеток, используемых в производстве конкретных препаратов, могут варьироваться в зависимости от вида животного-донора, используемого в качестве источника ткани, от возможного наличия посторонних агентов, природы препарата, его предполагаемых показаний к применению, особенностей производственного процесса и объема испытаний, проводимых на лекарственном препарате. Заявители должны объяснять и обосновывать подходы, использованные для конкретного препарата.
Глава 2. Оценка вирусной безопасности биологических
(биотехнологических) лекарственных средств, полученных
из клеточных линий человеческого и животного происхождения
1. Введение
В настоящей главе описаны испытания и оценка вирусной безопасности биологических (биотехнологических) лекарственных средств, полученных из охарактеризованных клеточных линий человеческого или животного происхождения (млекопитающих, птиц и насекомых), а также описаны данные, которые необходимо представлять в составе регистрационного досье, подаваемого в уполномоченные органы государств-членов при регистрации биологического лекарственного препарата. В настоящей главе в рамках понятия "вирус" не рассматриваются такие нетрадиционные трансмиссивные агенты, как возбудители губчатой энцефалопатии крупного рогатого скота и скрейпи овец и коз.
Положения настоящей главы относятся к лекарственным средствам, полученным с применением клеточных культур из охарактеризованных банков клеток. К ним относятся лекарственные препараты, полученные с применением клеточных культур in vitro, например, интерфероны, моноклональные антитела и препараты, полученные по технологии рекомбинантной ДНК, включая рекомбинантные субъединичные вакцины. К ним относятся также препараты, полученные при помощи гибридомной технологии in vivo из асцитической жидкости. К последнему случаю предъявляются особые требования, требования по проведению тестирования клеток из охарактеризованных банков, которые были впоследствии выращены in vivo, представлена в приложении N 1 к настоящей главе.
Положения настоящей главы применимы также к прочим биологическим средствам, частные требования к которым включают в себя ссылку на настоящую главу. Настоящая глава не распространяется на инактивированные вакцины, все живые вакцины, в том числе полученные методами генной инженерии.
Риск вирусной контаминации является особенностью всех биотехнологических препаратов, получаемых из клеточных линий. Такая контаминация может быть результатом контаминации исходных клеточных линий (субстратов клеток) или случайной контаминации вирусом в течение процесса производства и может иметь серьезные клинические последствия. Предполагается, что безопасность данных препаратов в отношении вирусной контаминации может быть в разумной степени обеспечена применением программы тестирования на наличие вируса, а также оценкой эффективности удаления (элиминации) и инактивации вируса в процессе производства в соответствии с рекомендациями, изложенными ниже.
Разработано 3 основных взаимодополняющих подхода к контролю потенциальной вирусной контаминации биотехнологических продуктов:
отбор и испытание клеточных линий и другого сырья, включая компоненты среды, на отсутствие контаминации вирусами, которые могут быть инфекционными и (или) патогенными для человека;
оценка возможностей процесса производства по очистке от инфекционных вирусов;
испытания продукта на соответствующих этапах процесса производства на отсутствие контаминации инфекционными вирусами.
Всем испытаниям присуще характерное для всех количественных методик определения вирусов ограничение: способность обнаруживать низкое содержание вирусов зависит (со статистической точки зрения) от размера выборки. В связи с этим ни один метод как таковой не позволяет установить безопасность препарата. Достоверность отсутствия вирусов в готовом препарате во многих случаях основана не только на прямых методах определения их наличия, но также на подтверждении того, что процесс очистки способен элиминировать и (или) инактивировать вирусы.
Необходимые виды и объем испытаний на вирусы и исследований очистки от вирусов на различных стадиях производства зависит от различных факторов, которые необходимо учитывать на каждой стадии в индивидуальном порядке. К факторам, которые необходимо принимать во внимание, относятся степень характеристики и квалификации банка клеток, природа обнаруженных вирусов, состав питательных сред, методы культивирования, конструкции производственной площадки и оборудования, результаты испытаний на вирусы после культивирования клеток, способность процесса обеспечивать элиминацию вирусов, а также вид препарата и его предполагаемое клиническое применение.
Цель настоящей главы состоит в описании общих подходов к испытанию продукта на наличие (отсутствие) вируса, экспериментов по оценке эффективности очистки от вирусов, рекомендованного подхода к планированию испытаний на наличие вирусов и исследований по очистке от вирусов.
Производители должны использовать указания настоящей главы с учетом особенностей производимого препарата и применяемого процесса производства. Необходимо объяснить и обосновать подход, использованный производителями для построения стратегии обеспечения вирусной безопасности. В дополнение к представлению подробных данных в целях ускорения экспертизы уполномоченным органом государства-члена следует представить резюме по оценке вирусной безопасности. В такое резюме необходимо включить краткое описание всех аспектов исследований вирусной безопасности и стратегий, использованных для предотвращения контаминации вирусами, указанных в настоящей главе.
2. Потенциальные источники вирусной контаминации
Вирусная контаминация биотехнологических препаратов может происходить от первичного источника клеточных линий или при случайном привнесении вируса во время процесса производства.
2.A. Вирусы, которые могут обнаруживаться в ГБК
Клетки могут иметь латентную или персистирующую вирусную инфекцию (например, герпетическую) или быть заражены эндогенным ретровирусом, который может передаваться вертикально от одного поколения клеток к другому, поскольку вирусный геном встроен в клеточный. Такие вирусы могут экспрессироваться конститутивно или их экспрессия может проявиться спонтанно.
Вирусы могут попасть в ГБК следующими путями:
при получении клеточных линий от инфицированных животных;
при использовании вируса для создания клеточной линии;
при использовании таких контаминированных биологических реагентов, как компоненты сыворотки животных;
при контаминации во время работы с клетками.
2.B. Посторонние вирусы, которые могут быть привнесены
во время процесса производства
Вирусы могут быть занесены в лекарственный препарат разными путями, в том числе:
при использовании таких контаминированных биологических реагентов, как компоненты сыворотки животных;
при применении вируса для стимуляции экспрессии генов, кодирующих необходимый белок;
при использовании контаминированного материала, например, колонки для аффинной хроматографии моноклональных антител;
при использовании контаминированного вспомогательного вещества во время получения лекарственной формы;
при контаминации в процессе производства, а также во время работы с клетками и культуральными средами. Мониторинг параметров клеточной культуры может способствовать раннему выявлению потенциальной контаминации посторонними вирусами.
3. Квалификация (аттестация) клеточной линии:
испытание на наличие вирусов
Важной частью характеристики клеточной линии для использования ее в производстве биотехнологичного препарата является надлежащее испытание на наличие вирусов.
3.A. Рекомендуемые испытания на наличие вирусов
для ГБК, РБК и клеток предельного для производства
клеточного возраста in vitro
Примеры испытаний на наличие вирусов, которые необходимо проводить однократно для клеток разных уровней, включая ГБК, РБК и клетки предельного для производства клеточного возраста in vitro приведены в таблице 1.
Таблица 1
Примеры испытаний на наличие вирусов, которые необходимо
проводить однократно для клеток разных уровней
|
Испытания
|
ГБК
|
РБК <1>
|
Клетки с предельным возрастом in vitro <2>
|
|
Испытания на наличие ретровирусов и других эндогенных вирусов
|
|||
|
Инфицирующая способность
|
+
|
-
|
+
|
|
Электронная микроскопия <3>
|
+ <3>
|
-
|
+ <3>
|
|
Обратная транскриптаза <4>
|
+ <4>
|
-
|
+ <4>
|
|
Прочие вирус-специфичные тесты <5>
|
если применимо <5>
|
-
|
если применимо <5>
|
|
Испытания на наличие неэндогенных или посторонних вирусов
|
|||
|
Испытания in vitro
|
+
|
- <6>
|
+
|
|
Испытания in vivo
|
+
|
- <6>
|
+
|
|
Испытания выработки антител <7>
|
+ <7>
|
-
|
-
|
|
Прочие вирус-специфичные тесты <8>
|
+ <8>
|
-
|
-
|
--------------------------------
<1> В соответствии с разделом 3.A.2 настоящей главы.
<2> Клетки с предельным возрастом in vitro: клетки с предельным для производства клеточным возрастом in vitro (в соответствии с разделом 3.A.3 настоящей главы).
<3> Позволяет также выявить и другие агенты.
<4> Не является необходимым при обнаружении инфицирующей способности ретровируса.
<5> Используется для клеточных линий, о которых известно, что они были инфицированы такими агентами.
<6> Для первого РБК данное испытаний должно проводиться на клетках с предельным клеточным возрастом in vitro, полученных из этого РБК, для последующих РБК один тест in vitro и in vivo нужно проводить либо непосредственно на РБК, либо на клетках с предельным клеточным возрастом in vitro.
<7> Испытания продукции мышиных (MAP), крысиных (RAP), хомячьих (HAP) антител, которые обычно применяются для клеточных линий грызунов.
<8> Испытания для клеточных линий, полученных от человека или нечеловекообразных приматов, или других клеточных линий в зависимости от обстоятельств.
3.A.1. Главный банк клеток.
На клетках ГБК нужно проводить интенсивные исследования по выявлению эндогенной и неэндогенной вирусной контаминации. В отношении гетерогибридных клеточных линий, в которых один или более партнеров являются человекообразными или нечеловекообразными обезьянами, необходимо провести испытания на вирусы человекообразных и нечеловекообразных обезьян, поскольку вирусная контаминация таких клеток может представлять собой особую опасность.
Выявление неэндогенных вирусов должно включать инокуляционные испытания in vitro и in vivo и любые другие специальные методы, в том числе такие видоспецифичные тесты, как тест продукции мышиных антител (MAP - mouse antibodies production), которые подходят для определения возможных контаминирующих вирусов с учетом истории пассажей клеточной линии.
3.A.2. Рабочий банк клеток.
Каждый РБК как исходный клеточный субстрат для производства биотехнологического продукта необходимо проверить на наличие посторонних вирусов либо непосредственно, либо путем проведения анализа клеток предельного для производства клеточного возраста in vitro, полученных из РБК. Если проводились испытания на наличие неэндогенных вирусов в РБК, а клетки, культивируемые до предельного для производства клеточного возраста in vitro или дольше, полученные из ГБК, были испытаны на наличие посторонних вирусов, сходные испытания на первоначальном РБК проводить не обязательно. Тесты образования антител на РБК обычно не проводят. Приемлем также альтернативный подход, в соответствии с которым полный набор исследований проводят в отношении РБК, а не ГБК.
3.A.3. Клетки предельного для производства клеточного возраста in vitro.
Предельный для производства клеточный возраст in vitro определяется на основании данных, полученных на клетках-продуцентах, выращенных в условиях опытно-промышленного или промышленного производства до предполагаемого предельного клеточного возраста in vitro или более. Клетки-продуценты получаются путем экспансии РБК, для их получения допускается также использовать ГБК. Клетки предельного клеточного возраста in vitro необходимо однократно проверить на наличие эндогенных вирусов, которые могли быть не выявлены в ГБК и РБК. По крайней мере, однократное испытание (in vitro и in vivo) клеток предельного для производства клеточного возраста in vitro позволяет убедиться в том, что процесс производства не подвержен контаминации посторонними вирусами. Если на этом этапе выявляются посторонние вирусы, процесс необходимо подвергнуть тщательной проверке для определения причины контаминации и при необходимости полностью реорганизовать его.
3.B. Рекомендуемые испытания для выявления
и идентификации вирусов
Для выявления эндогенных и посторонних вирусов могут использоваться разные методы количественного определения. В таблице 2 приведены примеры таких испытаний, в том числе все рекомендованные для использования протоколы количественного определения, однако этот перечень не является исчерпывающим или строго заданным.
Таблица 2
Примеры испытаний на наличие вирусов
|
Испытания
|
Исследуемый материал
|
Способность к обнаружению
|
Ограничения к обнаружению
|
|
Образование антител
|
лизат клеток и их культуральная среда
|
специфичные вирусные антигены
|
антигены не инфекционные для антигены животной тест-системы
|
|
Скрининг вируса in vivo
|
лизат клеток и их культуральная среда
|
широкий спектр вирусов, патогенных для человека
|
возбудители, которые не реплицируются или не вызывают заболеваний в тест-системах
|
|
Скрининг вируса in vitro для:
|
широкий спектр вирусов, патогенных для человека
|
возбудители, которые не способны к репликации или не вызывают поражений в тест-системах
|
|
|
1) характеристики Банка клеток
|
лизат клеток и их культуральная среда (при совместном культивировании исследуемый материал должен содержать интактные клетки)
|
||
|
2) скрининга производства
|
сбор необработанного продукта или лизат клеток и их культуральные среды из промышленного реактора
|
||
|
Трансмиссионная электронная микроскопия на:
|
вирус и вирусоподобные частицы
|
качественный анализ с оценкой идентичности
|
|
|
1) клеточном субстрате
|
жизнеспособные клетки
|
||
|
2) супернатанте клеточной культуры
|
бесклеточный супернатант культуры
|
||
|
Обратная транскриптаза (RT)
|
бесклеточный супернатант культуры
|
ретровирусы и экспресированная ретровирусная обратная транскриптаза
|
определяет только ферменты с оптимальной активностью при предпочтительных условиях, интерпретация может представлять сложность в связи с наличием клеточных ферментов, фона в некоторых концентрированных образцах
|
|
Инфицирующая способность ретровирусов (RV)
|
бесклеточный супернатант культуры
|
инфекционные ретровирусы
|
ретровирусы, не способные реплицироваться или не формирующие дискретные очаги или бляшки в выбранной тест-системе
|
|
Совместное культивирование
|
жизнеспособные клетки
|
инфекционные ретровирусы
|
ретровирусы, не способные реплицироваться
|
|
1) конечная точка инфицирующей способности
|
ретровирусы, не способные реплицироваться или не формирующие дискретные очаги или бляшки в выбранной тест-системе
|
||
|
2) конечная точка трансмиссионной электронной микроскопии
|
качественный анализ с оценкой идентичности кроме того, сложно отличить исследуемый материал от индикаторных клеток
|
||
|
3) конечная точка обратной транскриптазы
|
определяет только ферменты с оптимальной активностью при предпочтительных условиях. Интерпретация может представлять сложность в связи с наличием клеточных ферментов, фона в некоторых концентрированных образцах
|
||
|
ПЦР
|
клетки, культуральная жидкость и прочие материалы
|
специфичные последовательности вируса
|
должны присутствовать праймеры, не определяет инфицирующую способность вируса
|
Поскольку большинство используемых методик может изменяться с учетом научного прогресса, возможно использование альтернативных подходов при наличии данных, обосновывающих их применение. Производителям рекомендуется обсуждать такие альтернативные подходы с уполномоченными органами государств-членов. В отдельных ситуациях может потребоваться проведение других испытаний. Испытания должны включать в себя надлежащие контрольные материалы, гарантирующие достаточную чувствительность и специфичность. Во всех случаях, когда по виду животного - источнику происхождения клеточного субстрата - можно с относительно высокой вероятностью выявить наличие определенного вируса, может потребоваться проведение специальных испытаний и (или) применение других подходов. Если используемая в производстве клеточная линия получена от человекообразных или нечеловекообразных обезьян, необходимо дополнительно (при отсутствие должного обоснования) проверить ее на наличие таких вирусов человека, как вирусы, вызывающие иммунодефицита или гепатиты. Для выявления последовательностей нуклеиновых кислот, характерных для этих и других специфичных вирусов, может использоваться ПЦР. Далее представлено краткое описание общих принципов положений, в соответствии с которыми производитель должен обосновать свои действия.
3.B.1. Испытания на наличие ретровирусов.
При испытании клеток ГБК и клеток, культивируемых до предельного для производства клеточного возраста in vitro и дольше, следует проводить испытания на наличие ретровирусов, включая оценку инфицирующей способности на чувствительных клеточных культурах и электронную микроскопию. Если инфицирующая способность не выявлена и при помощи электронной микроскопии не были обнаружены ретровирусы или ретровирусоподобные частицы, проводятся исследования с использованием обратной транскриптазы или другие подходящие испытания на ретровирусы, которые могут не иметь инфицирующей способности. Исследования индукции в данном случае неэффективны.
3.B.2. Исследования in vitro.
Исследования in vitro проводятся путем инокуляции исследуемого материала, указанного в таблице 2, в разные восприимчивые индикаторные клеточные культуры, что позволяет определять большое число вирусов человека и животных. Выбор клеток для испытаний определяется видами животных, из которых получен подлежащий испытанию банк клеток, при этом необходимо включить клеточные линии человекообразных или нечеловекообразных обезьян, чувствительные к вирусам человека. Метод и материал для исследования выбираются в зависимости от типа вируса, наличие которого в материале предполагается с учетом происхождения клеток и проведенных с ними манипуляции. Необходимо проводить выявление как цитопатических, так и гемадсорбирующих вирусов.
3.B.3. Исследования in vivo.
Для выявления некультивируемых вирусов (не растущих в клеточных культурах) исследуемый материал, указанный в таблице 2, вводится животным, включая новорожденных и взрослых мышей, а также в развивающиеся эмбрионы птиц. В зависимости от происхождения исследуемых клеточных линий возможно проведение исследований на других видах животных. Следует контролировать состояние здоровья опытных животных, и любые отклонения от нормы нужно исследовать с целью установления причины заболевания.
3.B.4. Испытания на образование антител.
Видоспецифичные вирусы, присутствующие в клеточных линиях грызунов, можно выявить путем инокуляции исследуемого материала, указанного в таблице 2, не зараженным вирусом (безвирусным) животным с последующим определением сывороточных антител или ферментативной активности, проявляющихся спустя определенное время. В качестве примера можно привести тесты продукции мышиных (MAP), крысиных (RAP) и хомячьих (HAP) антител. Перечень вирусов, для выявления которых проводятся испытания продукции антител, приведен в таблице 3.
Таблица 3
Вирусы, для выявления которых
проводятся испытания продукции антител
|
Тест продукции мышиных (map) антител
|
Тест продукции хомячьих (hap) антител
|
Тест продукции крысиных (rap) антител
|
|
Вирус эктромелии <2>, <3>
|
Вирус лимфоцитарного хориоменингита (LCM) <1>, <3>
|
Вирус хантаан <1>, <3>
|
|
Вирус хантаан <1>, <3>
|
Вирус пневмонии мышей (PVM) <2>, <3>
|
Крысиный вирус Килхама (KRV) <2>, <3>
|
|
K-вирус <2>
|
Реовирус типа 3 (Reo) <1>, <3>
|
Вирус энцефаломиелита мышей (Theilers, GDVII) <2>
|
|
Вирус лактатдегидрогеназы (LDM) <1>, <3>
|
Вирус Сендай <1>, <3>
|
Вирус пневмонии мышей (PVM) <2>, <3>
|
|
Вирус лимфоцитарного хориоменингита (LCM) <1>, <3>
|
SV5
|
Коронавирус крыс (RCV) <2>
|
|
Мелкий вирус мышей <2>, <3>
|
Реовирус типа 3 (Reo) <1>, <3>
|
|
|
Аденовирус (MAV) <2>, <3>
|
Вирус Сендай <1>, <3>
|
|
|
Цитомегаловирус мышей (MCMV) <2>, <3>
|
Вирус сиалодакриоадеита (SDAV) <2>
|
|
|
Вирус энцефаломиелита мышей (Theilers, GDVII) <2>
|
Вирус Тоолан (HI) <2>, <3>
|
|
|
Вирус гепатита мышей (MHV) <2>
|
||
|
Ротавирус мышей (EDIM) <2>, <3>
|
||
|
Вирус пневмонии мышей (PVM) <2>, <3>
|
||
|
Полиомавирус <2>
|
||
|
Реовирус типа 3 (Reo3) <1>, <3>
|
||
|
Вирус Сендай <1>, <3>
|
||
|
Тимический вирус <2>
|
--------------------------------
<1> Вирусы, для которых доказана их способность инфицировать человека или приматов.
<2> Вирусы, для которых данные, доказывающие способность инфицировать человека, отсутствуют.
<3> Вирусы, способные реплицироваться in vitro в клетках человека или приматов.
3.C. Приемлемость клеточных линий
Некоторые клеточные линии, используемые для производства продукта, содержат эндогенные ретровирусы, другие вирусы или вирусные последовательности. Рекомендации по организации производства в таких случаях описаны в разделе 5 настоящей главы. Приемлемость клеточных линий, содержащих вирусы, не являющиеся эндогенными ретровирусами, определяется в индивидуальном порядке уполномоченными органами государств-членов с учетом анализа пользы и риска, исходя из пользы препарата и его предлагаемого клинического применения, свойств контаминирующих вирусов, их потенциала заражать человека или вызывать у него заболевания, процесса очистки препарата (например, анализ данных очистки от вирусов) и объема проведенных с очищенным нерасфасованным продуктом испытаний на вирусы.
4. Испытание необработанного нерасфасованного
продукта на наличие вирусов
Необработанный нерасфасованный продукт представляет собой один или несколько объединенных сборов клеток и культуральной среды. Если доступ к клеткам затруднен (например, при использовании полых волокон или аналогичных систем), то необработанный продукт может представлять собой жидкость, собранную из такого биореактора. Репрезентативный образец необработанного продукта, отобранного из промышленного биореактора перед дальнейшей обработкой, является одним из наиболее подходящих материалов, на котором можно с высокой вероятностью выявить контаминацию посторонними вирусами. Соответствующие испытания на наличие вирусов можно проводить на необработанном продукте, если только начальная частичная обработка не повышает чувствительность испытаний на вирусы (например, необработанный продукт может быть токсичным для исследуемых клеточных культур, в то время как частично обработанный продукт может быть нетоксичным).
В определенных случаях более приемлемым может быть анализ смеси, содержащей как интактные, так и разрушенные клетки и культуральные супернатанты, отобранные из промышленного биореактора перед последующей обработкой. В регистрационное досье необходимо включить данные, полученные как минимум на 3 сериях необработанного продукта, произведенного в опытно-промышленном или промышленном масштабе производства.
Производителям рекомендуется разрабатывать программы постоянной оценки на наличие посторонних вирусов в промышленных сериях продукта. Объем, количество и частота проведения испытаний на наличие вирусов в необработанном продукте определяется с учетом нескольких факторов: природы клеточных линий, используемых для производства требуемого лекарственного препарата, результатов и объема испытаний на наличие вирусов, проведенного во время квалификации клеточных линий, метода культивирования, источников сырья и результатов исследований по очистке от вирусов. В целях испытания необработанного продукта, как правило, проводятся скрининговые испытания in vitro с использованием одной или нескольких клеточных линий. При необходимости могут применяться методы на основе ПЦР или другие подходящие методы.
Собранный материал, в котором обнаружен посторонний вирус, не должен использоваться для производства продукта. Если на этом этапе выявляют посторонние вирусы, процесс производства необходимо тщательно проанализировать для определения причины контаминации и принятия соответствующих мер.
5. Обоснование и план действий при проведении исследований
по очистке от вирусов и испытаний на наличие вирусов
в очищенном нерасфасованном продукте
Необходимо разработать наиболее подходящий и рациональный протокол проведения испытаний на наличие вирусов начиная с ГБК на разных этапах производства вплоть до получения лекарственного препарата, включая оценку и характеристику эффективности очистки от вирусов необработанного продукта. Оценка и описание характеристик очистки от вирусов играют основную роль в данной схеме, их целью является получение надежного подтверждения того, что продукт не контаминирован вирусами.
При выборе вирусов, которые будут использоваться в исследовании по очистке от вирусов, целесообразно разделять необходимость анализа возможностей процессов очищать продукт от вирусов, о наличии которых известно, и потребность оценки устойчивости процесса путем описания очистки от неспецифических "модельных" вирусов. При оценке вирусной безопасности препарата для анализа процесса необходимо знать количество вирусов, которые могут содержаться в нем (например, в необработанном нерасфасованном продукте) и количество вирусов, которые можно инактивировать и элиминировать в процессе очистки. Знание временной зависимости процедур инактивации позволяет удостовериться в их эффективности. При анализе очистки от известных контаминантов требуются проведение детальных зависимых от времени исследований инактивации, подтверждение воспроизводимости инактивации и (или) элиминации и анализ параметров процесса. При описании процесса производства с точки зрения надежности очистки с использованием неспецифичных "модельных" вирусов в дизайне исследования необходимо уделить особое внимание безоболочечным вирусам. Объем исследований, необходимых для описания очистки от вирусов зависит от результатов испытания клеточных линий и необработанного продукта. Такие исследования необходимо проводить по методологии, предусмотренной разделом 6 настоящей главы.
Пример плана действий при проведении анализа процесса, описания очистки от вирусов и испытаний на вирусы очищенного продукта в зависимости от результатов испытаний клеток и (или) необработанного продукта на вирусы приведен в таблице 4.
Таблица 4
Пример плана действий при проведении процесса очистки
от вирусов и испытаний на вирусы в очищенном продукте
|
Ситуация
|
|||||
|
Статус
|
1
|
2
|
3 <2>
|
4 <2>
|
5 <2>
|
|
Наличие вируса <1>
|
-
|
-
|
+
|
+
|
(+) <3>
|
|
Вирусоподобные частицы <1>
|
-
|
-
|
-
|
-
|
(+) <3>
|
|
Ретровирусоподобные частицы <1>
|
-
|
+
|
-
|
-
|
(+) <3>
|
|
Выявлен вирус
|
не применимо
|
+
|
+
|
+
|
-
|
|
Вирус, патогенный для человека
|
не применимо
|
- <4>
|
- <4>
|
+
|
неизвестно
|
|
Действия
|
|||||
|
Характеристика процесса очистки от вирусов при использовании неспецифичных "модельных" вирусов
|
да <5>
|
да <5>
|
да <5>
|
да <5>
|
да <7>
|
|
Оценка процесса очистки от вирусов при использовании "релевантных" или специфичных "модельных" вирусов
|
нет
|
да <6>
|
да <6>
|
да <6>
|
да <7>
|
|
Испытания на наличие вируса в очищенном продукте
|
не применимо
|
да <8>
|
да <8>
|
да <8>
|
да <8>
|
--------------------------------
<1> Результаты испытаний на наличие вируса на уровне клеточного субстрата и (или) необработанного продукта. Как правило, не допускается использовать в производстве контаминированные вирусом клеточные культуры. Однако допускается использовать эндогенные вирусы (например, ретровирусы) или вирусы, являющиеся интегральной (составной) частью ГБК, если проведены надлежащие процедуры оценки вирусной очистки.
<2> Использование в качестве источника материала, контаминированного вирусами (независимо от их инфекционности и (или) патогенности для человека), допускается в исключительных случаях.
<3> Вирус обнаружен с помощью прямых или непрямых методов.
<4> Считающийся непатогенным.
<5> Необходимо провести описание очистки с помощью неспецифичных "модельных" вирусов.
<6> Необходимо провести оценку процесса с помощью "релевантных" или специфичных "модельных" вирусов.
<7> См. описание ситуации E.
<8> Необходимо подтвердить с помощью подходящих методов, обладающих высокой специфичностью и чувствительностью обнаружения искомого вируса, что в очищенном продукте вирусы не обнаруживаются. В регистрационном досье необходимо представить данные, по меньшей мере, о трех сериях необработанного продукта, полученного опытно-промышленным или промышленным способом. Однако определять наличие неинфекционных частиц в очищенном продукте из клеточных линий (например, клетки CHO), эндогенные частицы которых хорошо описаны и налажена их очистка, как правило, не требуется.
Далее рассматриваются различные варианты такого плана действий. Во всех случаях необходимо провести описание очистки с использованием неспецифичных "модельных" вирусов. Наиболее частыми являются ситуации 1 и 2. Клетки-продуценты, контаминированные вирусом, не являющимся ретровирусом грызунов, как правило, не используются. Если имеются убедительные и должным образом обоснованные основания для производства препаратов с использованием клеточной линии, описанной в ситуациях 3, 4 и 5, их необходимо согласовать с уполномоченным органом государства-члена. В случаях 3, 4 и 5 необходимо предусмотреть валидированные эффективные этапы, направленные на инактивацию и (или) элиминацию обнаруженного вируса в процессе производства.
Ситуация 1. Если в клетках и необработанном продукте вирусы, вирусоподобные частицы и ретровирусоподобные частицы не обнаружены, необходимо провести исследования элиминации и инактивации вирусов с использованием неспецифичных "модельных" вирусов.
Ситуация 2. Если обнаружены лишь ретровирусы грызунов или ретровирусоподобные частицы, которые считаются непатогенными (например, частицы A- и R-типов грызунов), необходимо провести анализ процесса с использованием специфичных "модельных" вирусов, например, вируса лейкоза мышей. Испытание очищенного продукта необходимо проводить с использованием подходящих методов, обладающих высокой специфичностью и чувствительностью в отношении рассматриваемого вируса. В регистрационном досье необходимо представить данные по меньшей мере о 3 сериях очищенного продукта, полученных опытно-промышленным или промышленным способом. В качестве субстратов для производства препаратов часто используются такие клеточные линии, как CHO, C127, BHK и клеточные линии гибридомы мышей, в отношении которых данные о проблемах с безопасностью, обусловленных вирусной контаминацией препаратов, не поступали. В отношении клеточных линий, эндогенные частицы которых хорошо описаны и очистка которых налажена, определение наличия неинфекционных частиц в очищенном продукте, как правило, не требуется. Необходимо провести исследования с использованием неспецифичных "модельных" вирусов, как описано в ситуации 1.
Ситуация 3. Если клетки или необработанный продукт содержат вирусы (за исключением ретровирусов грызунов), в отношении которых неизвестна их способность инфицировать человека (например, вирусы, указанные в сноске 2 к таблице 3 настоящей главы, за исключением ретровирусов грызунов (ситуация 2)), в исследованиях по изучению элиминации и инактивации вирусов необходимо использовать обнаруженный вирус. Если обнаруженный вирус использовать невозможно, в целях подтверждения приемлемости очистки необходимо использовать "релевантный" или специфичный "модельный" вирус. Зависимая от времени инактивация идентифицированных (или "релевантных", или специфичных "модельных") вирусов на критических стадиях инактивации должна стать частью анализа процесса на предмет наличия таких вирусов. Испытание очищенного продукта необходимо проводить с использованием подходящих методов, обладающие высокой специфичностью и чувствительностью в отношении рассматриваемого вируса. В регистрационном досье необходимо представить данные по меньшей мере о 3 сериях очищенного продукта, полученных опытно-промышленным или промышленным способом.
Ситуация 4. Если обнаружен известный патоген человека (например, описанный в сноске 1 к таблице 3 настоящей главы), использовать продукт допускается в исключительных случаях. В связи с этим в исследованиях по изучению элиминации и инактивации вирусов рекомендуется применять обнаруженный вирус с использованием подходящих методов, обладающих высокой специфичностью и чувствительностью в отношении его. Если обнаруженный вирус использовать невозможно, необходимо использовать "релевантный" и (или) специфичный "модельный" вирус. Необходимо подтвердить способность процесса в ходе очистки и инактивации элиминировать и инактивировать отобранные вирусы. В ходе анализа процесса на ключевых этапах инактивации необходимо получить данные о зависимой от времени инактивации. Испытание очищенного продукта необходимо проводить с использованием подходящих методов, обладающих высокой специфичностью и чувствительностью в отношении рассматриваемого вируса. В регистрационном досье необходимо представить данные по меньшей мере о 3 сериях очищенного продукта, полученных опытно-промышленным или промышленным способом.
Ситуация 5. Если в клетках или необработанном продукте обнаруживается вирус, который невозможно классифицировать с помощью имеющихся методов, продукт, как правило, считается непригодным, поскольку вирус может быть патогенным. В очень редких случаях при наличии убедительных и обоснованных причин для производства препарата с использованием такой клеточной линии перед продолжением процесса производства необходимо согласование с уполномоченным органом государства-члена.
6. Оценка и характеристика процедур очистки от вирусов
Анализ и описание процедур элиминации и (или) инактивации вирусов играют важную роль в обеспечении безопасности биологических (биотехнологических) препаратов. Контаминация биологических препаратов агентами, о наличии которых не было известно и это не предполагалось, происходила если эти препараты были получены из не описанных всесторонне клеточных линий. Оценка очистки от вирусов обеспечивает определенную уверенность в том, что все неизвестные, неподозреваемые и опасные вирусы будут элиминированы. Исследования следует проводить с надлежащим документированием и контролем.
Целями исследований по очистки от вирусов являются оценка этапов процесса производства, которые можно считать эффективными для инактивации (элиминации) вирусов, и количественная оценка суммарного снижения содержания вирусов в продукте с помощью этих этапов. Это достигается за счет преднамеренного одномоментного добавления значимого количества вирусов в необработанный материал и (или) к различным фракциям, получаемым на различных этапах процесса производства, и подтверждения того, что на последующих этапах достигается их необходимая элиминация или инактивация. Если надлежащая очистка достигается за счет меньшего количества этапов, то оценивать и описывать с точки зрения оценки вирусной безопасности остальные этапы процесса производства не требуется. Следует помнить, что остальные этапы процесса производства могут косвенно влиять на достигнутую инактивацию (элиминацию). Производители должны объяснить и обосновать подход, использованный для проведения исследований по очистке от вирусов.
Снижение инфицирующей способности вирусов достигается за счет элиминации вирусных частиц или их инактивации. Необходимо описать возможные механизмы снижения инфицирующей способности вируса на каждом изученном этапе процесса производства и указать, достигнуты они за счет инактивации или элиминации. Исследование этапов инактивации необходимо спланировать таким образом, чтобы пробы отбирались в различные временные точки, при этом следует построить кривую инактивации в соответствии с подразделом 6.B.5 настоящей главы.
Исследования по оценке очистки от вирусов проводятся в целях подтверждения очистки от вирусов, которые содержатся в ГБК, и (или) обеспечения определенной степени уверенности в том, что посторонние вирусы, которые могли быть не выявлены или могли контаминировать процесс производства, были устранены. Факторы снижения вирусной нагрузки, как правило, выражаются в логарифмических координатах, то есть несмотря на то, что остаточная инфицирующая способность вируса никогда не будет снижена до нуля, ее можно значительно снизить с математической точки зрения.
В дополнение к исследованиям по очистке от вирусов, о наличии которых известно, необходимо провести исследования по оценке возможностей процесса элиминировать и (или) инактивировать другие вирусы. Цель исследований с использованием вирусов, которые обладают различными биохимическими и биофизическими свойствами, о содержании которых неизвестно или наличие которых не ожидается, заключается, как правило, в изучении надежности процедур, но не в обеспечении определенной степени их инактивации или элиминации (специфического риска). Желательно обеспечить подтверждение способности процесса производства инактивировать или элиминировать вирусы в соответствии с подразделом 6.C настоящей главы. Такие исследования не направлены на оценку специфического (определенного) риска безопасности, то есть не требуется достигать определенной степени очистки для данного риска.
6.A. Выбор вирусов для оценки и характеристики
элиминации вирусов
Чтобы испытать общую способность системы очищать продукт от вирусов, в исследования по оценке очистки и описанию процесса необходимо включать вирусы, напоминающие по свойствам вирусы, которые могут контаминировать препарат и обладают широким диапазоном физико-химических свойств. Руководствуясь настоящей главой, производитель должен обосновать выбор вирусов в соответствии с целями исследований по оценке и описанию и на основании настоящей главы.
6.A.1. "Релевантные" вирусы и "модельные" вирусы.
Основным аспектом проведения исследования по очистке от вирусов является определение, какие вирусы в него включать. Такие вирусы делятся на три категории: "релевантные" вирусы, специфичные "модельные" вирусы и неспецифичные "модельные" вирусы.
Необходимо подтвердить способность процесса очистки и (или) инактивации удалять и (или) инактивировать "релевантные" вирусы. Если "релевантного" вируса нет в наличии или он не адаптирован к исследованиям по оценке процесса очистки от вирусов (например, его невозможно вырастить в достаточно высоком титре in vitro), в качестве замены необходимо использовать специфичные "модельные" вирусы.
Клеточные линии, полученные от грызунов, обычно содержат частицы эндогенного ретровируса или ретровирусоподобные частицы, которые могут быть инфекционными (частицы C-типа) или неинфекционными (цитоплазматические частицы A- и R-типа). Необходимо определить способность процесса производства элиминировать и (или) инактивировать ретровирусы в продуктах, полученных с использованием таких клеток. Этого можно достичь с использованием вируса лейкемии мышей - специфичный "модельный" вирус для клеток мышиного происхождения. Например, при иммортализации B-лимфоцитов вирусом Эпштейна-Барр (EBV) и получении клеточных линий человека, секретирующих моноклональные антитела, необходимо определить способность процесса производства элиминировать и (или) инактивировать вирус герпеса. В качестве специфичного "модельного" вируса может также использоваться вирус псевдобешенства.
Если целью служит описание общей способности процесса производства элиминировать и (или) инактивировать вирусы (описание надежности процесса очистки), необходимо провести исследования по характеристике очистки с использованием неспецифичных "модельных" вирусов, обладающих различными свойствами. В таком анализе могут применяться результаты исследований с "релевантными" и (или) специфичными "модельными" вирусами. Проводить испытания со всеми типами вирусов не требуется. Необходимо отдавать предпочтение вирусам, которые проявляют значительную устойчивость к физическим и (или) химическим воздействиям. Результаты изучения таких вирусов служат источником доказательных сведений об общей способности процесса производства элиминировать и (или) инактивировать вирусы. Выбор и количество использованных вирусов зависят от качества и степени описания клеточных линий и процесса производства.
Примеры подходящих "модельных" вирусов, обладающих широким диапазоном физико-химических структур, и примеры вирусов, использованных в исследованиях по очистке от вирусов, представлены в приложении N 2 к настоящей главе.
6.A.2. Рассмотрение других аспектов.
Предпочтительно использовать вирусы, которые можно вырастить (обогатить) до высокого титра, однако это не всегда возможно.
Необходимо располагать эффективной и надежной методикой количественного определения каждого использованного вируса на каждом исследуемом этапе производства.
Необходимо учитывать потенциальный вред здоровью, который может быть нанесен персоналу, проводящему исследования очистки.
6.B. Дизайн и обязательные условия проведения исследований
по оценке и описанию характеристик очистки вирусов
6.B.1. Производственные помещения, оборудование и персонал.
В соответствии с правилами надлежащей производственной практики Союза, утверждаемыми Комиссией, вносить какие-либо вирусы в производственное оборудование не допускается. В связи с этим исследования по очистке от вирусов должны проводиться в отдельной лаборатории, оборудованной для работы с вирусами, персоналом с вирусологической квалификацией в сотрудничестве с производственным персоналом, занимавшимся планированием и подготовкой уменьшенной версии процесса очистки.
6.B.2. Система производства в уменьшенном масштабе.
Необходимо доказать валидность (пригодность) уменьшенного масштаба производства. Уровень очистки в уменьшенном масштабе должен максимально соответствовать методу производства. Необходимо подтвердить, что все параметры (хроматографическое оборудование, высота слоя сорбента колонки (column bed-height), линейная скорость потока (linear flow-rate), отношение скорости потока к объему слоя сорбента (flow-rate-to-bed-volume ratio) (время контакта), виды буферных растворов и гелей, значение pH, температура и концентрация белка, соли и продукта) максимально приближены к промышленному способу производства. Необходимо получить тот же профиль элюирования. В отношении процедур необходимо соблюдать аналогичные требования. Неизбежные отклонения необходимо проанализировать с точки зрения их влияния на результаты.
6.B.3. Анализ поэтапной элиминации вирусов.
При проведении исследований по очистке от вирусов желательно оценить вклад более чем одного этапа производства в элиминацию вирусов. Этапы, на которых, вероятнее всего, устраняются вирусы, необходимо отдельно оценить на предмет их способности элиминировать и инактивировать вирусы, при этом следует уделить особое внимание точному разграничению отдельных этапов. На каждом подлежащем испытанию этапе материал должен содержать достаточное количество вируса, необходимое для оценки эффективности каждого этапа. Как правило, на каждом подлежащем испытанию этапе вирус добавляется во внутрипроизводственный материал. В некоторых случаях, достаточно просто добавить вирус в высоком титре в необработанный продукт и измерять его концентрацию между последовательными этапами. Если элиминация вируса достигается с помощью процедур отделения, рекомендуется при необходимости и по возможности изучить распределение вирусной нагрузки по различным фракциям. Если на множестве этапов процесса производства используются вирулицидные буферные растворы, как часть оценки всего процесса допускается использовать альтернативные стратегии, например, параллельное добавление менее вирулицидных буферных растворов. Титр вируса необходимо определять до и после каждого исследуемого этапа. В целях обеспечения необходимой статистической значимости результатов необходимо использовать методики количественного определения инфицирующей способности с высокой чувствительностью и воспроизводимостью с достаточным числом повторностей. При достаточном обосновании допускается использовать количественные методики, не направленные на выявление инфицирующей способности. В целях обеспечения чувствительности методов во все методики по определению инфицирующей способности необходимо включать надлежащий вирусный контроль. К тому же необходимо принимать во внимание особенности отбора проб вируса в низких концентрациях в соответствии с требованиями к статистическим подходам к интерпретации результатов испытаний на вирусы согласно приложению N 3 к настоящей главе.
6.B.4. Определение вклада физической элиминации и инактивации вирусов.
Снижение инфицирующей способности вируса достигается за счет его элиминации или инактивации. Необходимо описать возможные механизмы снижения инфицирующей способности вируса на каждом изученном этапе процесса производства и указать, достигнуты они за счет инактивации или элиминации. Если процесс производства не позволяет добиться удовлетворительного снижения инфицирующей способности, а элиминация вируса рассматривается в качестве основного фактора безопасности препарата, необходимо внедрить специальные или дополнительные этапы инактивации и (или) элиминации. На определенном этапе может потребоваться разграничить элиминацию и инактивацию (например, если есть вероятность того, что буферный раствор, использованный более чем на одном этапе, может влиять на инактивацию на каждом этапе, то есть влияние буферного раствора на инактивацию распределяется между несколькими хроматографическими этапами, при этом необходимо определить степень элиминации, достигаемую за счет каждого из этих хроматографических этапов).
6.B.5. Оценка инактивации.
Для оценки инактивации вируса в необработанном продукте или промежуточном материале должен быть введен инфекционный вирус и рассчитан фактор (коэффициент) снижения вирусной нагрузки. Следует учитывать, что инактивация вируса не является простым процессом первого порядка, обычно она более сложная и имеет быструю фазу 1 и медленную фазу 2. Именно поэтому исследование должно быть спланировано так, чтобы отбор проб проводился в разные временные точки, и была построена кривая инактивации. В исследования по инактивации помимо временной точки, соответствующей минимальной экспозиции, рекомендуется включать не менее одной временной точки, которая предшествует точке минимальной экспозиции и отличается от ноля. Дополнительные данные особенно необходимы, если "релевантный" вирус является патогенным для человека и создан процесс эффективной его инактивации. Однако в исследованиях инактивации, в которых неспецифичные "модельные" вирусы или специфичные "модельные" вирусы используются в качестве суррогатов вирусных частиц (например, внутрицитоплазматические ретровирусоподобные частицы в клетках яичника китайского хомячка) воспроизводимость очистки необходимо подтвердить по меньшей мере в 2 исследованиях. Начальную вирусную нагрузку по возможности необходимо определять путем выявления вируса после его добавления в исходный материал. Если это невозможно, то начальная вирусная нагрузка рассчитывается по титру вируса в добавляемом к исходному материалу препарате. Если высокая скорость инактивации не позволяет построить кривую инактивации с использованием условий процесса, необходимо предусмотреть надлежащие контроли, в целях подтверждения того, что в ходе инактивации инфицирующая способность была устранена.
6.B.6. Использование и регенерация хроматографических колонок.
Со временем или при повторном (многократном) использовании хроматографических колонок и других систем, используемых в процессе очистки, их способность к элиминации вируса может изменяться. Определение стабильности очистки от вирусов после многократного применения оправдывает возможность повторного использования колонок. Необходимо подтвердить, что вирусы, которые были потенциально задержаны производственной системой, должным образом уничтожены и удалены перед повторным ее использованием. Таким подтверждением, к примеру, может служить демонстрация того, что процедуры очистки и регенерации инактивируют или элиминируют вирус.
6.B.7. Особые меры предосторожности.
При приготовлении вирусов в высоком титре необходимо соблюдать осторожность, чтобы не допустить их агрегации, которая может улучшить физическую элиминацию, но снизить инактивацию и исказить таким образом корреляцию с фактическим производством.
Необходимо учитывать минимальное количество вируса, содержание которого можно достоверно определить.
В целях анализа снижения инфицирующей способности вируса вследствие разведения, концентрации, фильтрации или хранения проб перед их разведением, в исследование необходимо включать параллельные контрольные методики количественного определения.
"Добавление" вирусов необходимо осуществлять в небольшом объеме, чтобы не разводить продукт или не изменить его свойства. Проба испытуемого белка после его разведения больше не является идентичной продукту, получаемому промышленным способом.
Небольшие различия в буферных растворах, питательной среде, реактивах и т.п. могут значительно повлиять на очистку от вирусов.
Инактивация вирусов зависит от времени, поэтому время, в течение которого продукт, в который добавлен вирус, остается в определенном буферном растворе или определенной хроматографической колонке, должно соответствовать условиям промышленного процесса производства.
Буферные растворы и продукт необходимо оценивать отдельно на токсичность и влияние на результаты методик, применяемых для определения титра вируса, так как эти компоненты могут негативно влиять на индикаторные клетки. Если такие растворы токсичны для индикаторных клеток, могут потребоваться разведение, коррекция pH или диализ буферного раствора, содержащего добавленный вирус. Если продукт обладает собственной противовирусной активностью, может потребоваться проведение исследования очистки без продукта в "имитационном" цикле ("mock" run), хотя при этом исключение продукта или замена его на аналогичный белок, не обладающий противовирусной активностью, может повлиять на поведение вирусов на некоторых этапах производства. Необходимо включить достаточные контроли для оценки влияния процедур, используемых исключительно для приготовления проб для методик определения (например, диализ, хранение), на элиминацию и (или) инактивацию добавленного вируса.
Во многих схемах очистки повторно используются одинаковые или схожие буферные растворы или колонки. При анализе данных это необходимо принимать во внимание. Эффективность конкретного метода элиминации вируса может зависеть от этапа производства, на котором он используется.
Если условия производства или буферные растворы чрезмерно цитотоксичны или вирулицидны, совокупные факторы (коэффициенты) снижения вирусной нагрузки могут быть недооценены, в этом случае оценка проводится в индивидуальном порядке. Ввиду внутренних ограничений или несоответствующего дизайна исследований по очистке от вирусов общие факторы (коэффициенты) снижения вирусной нагрузки могут быть также переоценены.
6.C. Интерпретация результатов исследований
по очистке от вирусов
Целями оценки инактивации и (или) элиминации вирусов являются анализ и описание этапов процесса производства, считающихся эффективными для инактивации и (или) элиминации вирусов, и количественное определение общего снижения содержания вирусов, достигаемого при производстве. При наличии вирусных контаминантов (ситуации 2 - 5), чтобы обеспечить надлежащий уровень безопасности лекарственного препарата, необходимо не только подтвердить, что вирус элиминирован или инактивирован, но и показать, что в процессе очистки от вирусов остается запас эффективности. Количество элиминированного или инактивированного в ходе процесса производства вируса необходимо сравнить с количеством, которое содержалось в необработанном продукте.
Для осуществления такого сравнения необходимо оценить содержание вируса в необработанном продукте. Такая оценка осуществляется с использованием методик определения инфицирующей способности или иных методик, например, трансмиссионной электронной микроскопии (ТЭМ). Весь процесс очистки должен элиминировать значительно большее количество вируса, чем его содержание в объеме необработанного продукта, эквивалентном одной дозе лекарственного препарата. Расчет факторов (коэффициентов) снижения вирусной нагрузки в исследованиях по очистке от вирусов осуществляется согласно приложению N 4 к настоящей главе, расчет ожидаемого количества частиц на дозу - согласно приложению N 5 к настоящей главе.
Механизмы очистки от различных классов вирусов могут отличаться. При анализе данных, подтверждающих эффективность процедур инактивации и (или) элиминации вирусов, необходимо использовать комбинацию факторов:
пригодность использованных тест-вирусов;
дизайн исследований очистки;
достигнутое снижение вирусной нагрузки (в логарифмах);
зависимость инактивации от времени;
потенциальное влияние изменения параметров процесса производства на инактивацию и (или) элиминацию вируса;
чувствительность (пределы определения и обнаружения) аналитических методик;
селективность процедур инактивации и (или) элиминации в отношении определенных классов вирусов.
Эффективная очистка достигается любым из следующих способов: многоэтапная инактивация, многоэтапное дополнительное отделение или комбинация этапов инактивации и отделения. Отделение "модельных" вирусов может происходить иначе, нежели отделение искомого вируса, поскольку методы отделения могут зависеть от высокоспецифичных физико-химических свойств вируса, которые влияют на их взаимодействие с гелевыми матрицами и на свойства преципитации. Параметры производства, влияющие на отделение, подлежат надлежащему описанию и контролю. Различия могут быть обусловлены изменением поверхностных свойств, например, гликозилированием. Однако, несмотря на такие потенциальные различия, эффективная элиминация достигается путем комбинации дополнительных этапов отделения или комбинации этапов инактивации и отделения. Таким образом, хорошо спланированные этапы отделения, например, хроматографические процедуры, фильтрация и экстракция, могут оказаться эффективными способами элиминации вирусов, если они осуществляются в хорошо контролируемых условиях. Эффективный этап элиминации вирусов должен обладать воспроизводимостью в отношении снижения вирусной нагрузки, которую необходимо подтвердить не менее чем двумя независимыми исследованиями.
Совокупный фактор (коэффициент) снижения, как правило, выражается в виде суммы отдельных факторов. Если индивидуальный фактор (коэффициент) снижения составляет 1,0 lg или менее, то он признается незначительным и при отсутствии обоснования не учитывается в расчете совокупного фактора (коэффициента) снижения.
Если процесс производства не позволяет добиться удовлетворительного снижения инфицирующей способности, а элиминация вируса рассматривается в качестве основного фактора безопасности препарата, необходимо внедрить дополнительные этапы инактивации и (или) элиминации. В отношении всех вирусов производители должны обосновать приемлемость полученных факторов (коэффициентов) снижения. Результаты должны быть оценены на основании указанных факторов.
6.D. Ограничения исследований по очистке от вирусов
Исследования по очистке от вирусов влияют на обеспечение приемлемого уровня безопасности лекарственного препарата, однако сами по себе не определяют его безопасность. Тем не менее ряд элементов дизайна и выполнение исследований по очистке от вирусов могут привести к некорректной оценке способности процесса снижать инфицирующую способность вирусов. К таким факторам относятся следующие:
Вирусные препараты, использованные в исследованиях по очистке, как правило, получают на культуре тканей. Поведение вируса из культуры ткани на этапе производства может отличаться от свойств природного вируса, например, если природный и выращенный вирусы различаются по чистоте или степени агрегации.
Инактивация инфицирующей способности вирусов, как правило, описывается двухфазной кривой, в которой быстрая начальная фаза сменяется медленной. Существует вероятность, что вирусы, оставшиеся после первого этапа инактивации, могут быть более устойчивыми к последующим этапам. Например, если резистентная фракция принимает форму вирусных агрегатов, их инфицирующая способность может быть устойчива к различным химическим воздействиям и нагреванию.
Возможность совокупного процесса снижать инфицирующую способность выражается в виде суммы логарифмов снижения вирусной нагрузки на каждом этапе. Суммирование факторов (коэффициентов) снижения на различных этапах, особенно этапах с небольшим снижением (например, менее 1,0 lg), может привести к завышению истинного показателя оценки элиминации вируса. Кроме того, не допускается суммировать факторы (коэффициенты) снижения, полученные за счет повторения идентичных или схожих процедур, без должного основания.
Выражение факторов (коэффициентов) снижения в виде логарифма снижения титра означает, что, несмотря на существенное снижение остаточной инфицирующей способности вируса, она никогда не снизится до нуля. Например, снижение инфицирующей способности препарата, содержащего 8,0 lg инфицирующих единиц на миллилитр, на фактор 8,0 lg дает 0,0 lg на миллилитр или одну инфицирующую единицу на миллилитр с учетом предела обнаружения методики.
Опытно-промышленный способ обработки может отличаться от промышленного, даже несмотря на дизайн процесса с уменьшенным масштабом.
Сложение индивидуальных факторов (коэффициентов) снижения вирусной нагрузки, достигнутых за счет схожих механизмов инактивации, может привести к переоценке совокупной очистки от вирусов.
6.E. Статистический анализ данных
В целях интерпретации результатов исследований очистки от вирусов они должны включать в себя статистический анализ данных. Для подтверждения полученных выводов результаты исследования должны быть статистически значимы в соответствии с приложением N 3 к настоящей главе.
6.F. Повторная оценка очистки от вирусов
При значимых изменениях процесса производства или очистки необходимо оценить прямое и косвенное их влияние на очистку от вирусов и при необходимости подвергнуть систему повторной оценке. Например, изменения процессов производства могут служить причиной изменения продукции вирусов клеточной линией, изменения этапов производства могут повлиять на степень очистки от вирусов.
7. Заключение
В настоящей главе охарактеризованы подходы к оценке рисков вирусной контаминации и очистке от вирусов продукта, которые вносят вклад в производство безопасных биотехнологических препаратов, получаемых из клеточных линий человека или животных и описано значение ряда стратегий, включая:
тщательное описание (скрининг) исходного материала клеточного субстрата на предмет наличия вирусных контаминантов;
оценку риска путем выявления контаминантов, тропных к организму человека;
внедрение надлежащей программы испытаний на посторонние вирусы в необработанном продукте;
детальное планирование исследований очистки от вирусов с использованием различных методов инактивации или элиминации вирусов в одном и том же процессе производства с целью достижения максимальной очистки от вирусов;
проведение исследований, направленных на анализ инактивации и элиминации вирусов.
Приложение N 1
к главе 2 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ
К ПРЕПАРАТАМ ПОЛУЧЕННЫМ ИЗ ОХАРАКТЕРИЗОВАННЫХ БАНКОВ
КЛЕТОК, КОТОРЫЕ БЫЛИ ВПОСЛЕДСТВИИ ВЫРАЩЕНЫ IN VIVO
Для препаратов, производимых из жидкостей, полученных от животных, инокулированных клетками охарактеризованных банков клеток, необходимо представлять дополнительные сведения о животных.
Если возможно, животных, используемых при производстве биотехнологических (биологических) продуктов, необходимо отбирать из четко охарактеризованных беспатогенных колоний. Должно быть проведено полноценное исследование на наличие вирусов (например, вирусов, указанных в таблице 3 главы 2 настоящих Правил). Должны быть описаны карантинные мероприятия для вновь прибывших и заболевших животных, а также должна обеспечиваться достаточность всех методов изоляции, очистки и деконтаминации, применяемых в питомнике, для сдерживания распространения посторонних агентов. Это можно сделать при использовании программы-сигнализатора. Необходимо представить перечень агентов, в отношении которых проводятся испытания. В питомнике или в непосредственной близости к нему должна быть организована ветеринарная служба. Необходимо описать, насколько хорошо виварий изолирован от других отделов производственного объекта. Методы, используемые персоналом в процессе производства препарата, должны быть достаточны для обеспечения вирусной безопасности.
Необходимо полностью описать процедуры содержания животных, включая рацион, график уборки и кормления, положение о периодическом ветеринарном наблюдении, если применимо, а также сведения о содержании животных, которым инокулирован возбудитель. Необходимо также представить описание режимов предварительной иммунизации животных, подготовки инокулята, а также сведения о точке и методе инокуляции.
Первичный собранный материал от животных может рассматриваться в качестве эквивалента необработанного продукта, полученного из биореактора. Именно поэтому к нему применяются все положения для испытаний, указанные в пункте 4 главы 2 настоящих Правил. В дополнение к этому производитель должен оценить бионагрузку в необработанном продукте, определить, свободен ли материал от микоплазм, и провести видоспецифичные пробы, а также испытания in vivo на взрослых и новорожденных мышах.
Приложение N 2
к главе 2 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ПЕРЕЧЕНЬ
ВИРУСОВ, ИСПОЛЬЗУЕМЫХ В ИССЛЕДОВАНИЯХ ПО ОЧИСТКЕ ОТ ВИРУСОВ
I. Полезные "модельные" вирусы
В качестве неспецифичных "модельных" вирусов, обладающих широким набором физико-химических структур используются:
SV40 (полиомавирус макак 1), вирус полиомиелита человека 1 (Сэбина), парвовирус животных или некоторые другие некрупные безоболочечные вирусы;
вирусы гриппа и парагриппа, вирус Синдбис или другие безоболочечные РНК-вирусы от средних до крупных размеров;
вирусы герпеса (например, ВПГ-1 или вирус псевдобешенства) или некоторые другие ДНК-вирусы от средних до крупных размеров.
Указанные вирусы являются лишь примерами, их использование необязательно.
В качестве специфичных в отношении клеточных линий грызунов "модельных" вирусов используются ретровирусы мышей.
II. Вирусов, используемые в исследованиях
по очистке от вирусов
Некоторые вирусы, используемые в исследованиях по очистке от вирусов, перечислены в таблице A-1. Поскольку это исключительно примерный перечень и использование каких-либо вирусов из этой таблицы необязательно, производители вправе предлагать другие вирусы, особенно если они лучше подходят для отдельного процесса производства. Необходимо, как правило, проанализировать способность процесса очищать продукт по меньшей мере от трех различных вирусов с различными характеристиками.
Таблица A-1
Вирусы, используемые в исследованиях по очистке от вирусов
|
Вирус
|
Семейство
|
Род
|
Естественный хозяин
|
Геном
|
Оболочка
|
Размер (нм)
|
Форма
|
Резистентность <*>
|
|
Вирус везикулярного стоматита
|
рабдовирусы
|
везикуловирус
|
лошадь, корова
|
РНК
|
да
|
70 x 150
|
пуля
|
низкая
|
|
Вирус парагриппа
|
парамиксо-вирусы
|
парамиксо-вирус
|
различные
|
РНК
|
да
|
100 - 200
|
плеосфера
|
низкая
|
|
Вирус лейкемии мышей (MuLV)
|
ретровирусы
|
онковирус c типа
|
мышь
|
РНК
|
да
|
80 - 110
|
сферическая
|
низкая
|
|
Вирус Синдбис
|
тогавирусы
|
альфавирус
|
человек
|
РНК
|
да
|
60 - 70
|
сферическая
|
низкая
|
|
Вирус вирусной диареи коров (BVDV)
|
флавивирусы
|
пестивирус
|
корова
|
РНК
|
да
|
50 - 70
|
плеосфера
|
низкая
|
|
Вирус псевдобешенства
|
герпесвирусы
|
свинья
|
ДНК
|
да
|
120 - 200
|
сферическая
|
средняя
|
|
|
Вирус полиомиелита 1 типа
|
пикорнавирусы
|
энтеро-вирус
|
человек
|
РНК
|
нет
|
25 - 30
|
икосаэдрическая
|
средняя
|
|
Вирус энцефало-миокардита (EMC)
|
пикорнавирусы
|
кардио-вирус
|
мышь
|
РНК
|
нет
|
25 - 30
|
икосаэдрическая
|
средняя
|
|
Реовирус 3
|
реовирусы
|
орторео-вирус
|
различные
|
РНК
|
нет
|
60 - 80
|
сферическая
|
средняя
|
|
SV40
|
паповавирусы
|
полиома-вирус
|
обезьяна
|
ДНК
|
нет
|
40 - 50
|
икосаэдрическая
|
очень высокая
|
|
Парвовирусы (собак, свиней)
|
парвовирусы
|
парвовирус
|
собака, свинья
|
ДНК
|
нет
|
18 - 24
|
икосаэдрическая
|
очень высокая
|
--------------------------------
<*> Резистентность к физико-химическим воздействиям основана на исследованиях процессов производства. Резистентность относительна к специфичному воздействию и используется для понимания биологии вируса и характера процесса производства. Фактические результаты зависят от вида воздействия.
Приложение N 3
к главе 2 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ
К СТАТИСТИЧЕСКИМ ПОДХОДАМ ИНТЕРПРЕТАЦИИ РЕЗУЛЬТАТОВ
ИСПЫТАНИЯ НА ВИРУСЫ
1. При определении титра вирусов возникают те же проблемы, что и для всех биологических методов анализа. Чтобы определить достоверность исследования, необходимо проанализировать правильность (accuracy) определения вирусных титров и факторов (коэффициентов) снижения вирусной нагрузки, полученных на их основании, и провести валидацию методик. Целью статистического анализа является подтверждение проведения исследования с приемлемым уровнем компетентности в отношении вирусологических данных.
2. Аналитические методики могут быть качественными и количественными. К качественным методикам относятся методики определения инфицирующей способности на животных и методики инфекционной дозы на культуре ткани (TCID), в которых подсчитывается количество инфицированных и неинфицированных животных или клеток. Затем по доле инфицированных животных или клеток определяются инфицирующие титры. В количественных методиках определяемая инфекционность зависит от вирусной нагрузки. К количественным методикам относятся методики бляшкообразования, в которых каждая бляшка принимается за одну инфекционную единицу, ПЦР в режиме реального времени. Результаты применения как качественных, так и количественных аналитических методик подлежат статистической обработке.
3. Вариабельность результатов методик может быть обусловлена ошибками при разведении, статистическими (случайными) эффектами и различиями в аналитических системах, которые либо неизвестны, либо трудно поддаются контролю. При сравнении результатов различных аналитических циклов (вариация между циклами (between-assay variation)) величина таких эффектов оказывается выше результатов внутри одного цикла (вариация внутри цикла (within-assay variation)).
4. Границы 95% доверительных интервалов результатов внутри одного цикла должны, как правило, укладываться в интервал  0,5 lg от среднего. Вариабельность внутри цикла оценивается с помощью стандартных статистических тестов. Вариабельность между циклами можно определить путем включения препарата сравнения, значение активности (potency) которого должно укладываться в
0,5 lg от среднего. Вариабельность внутри цикла оценивается с помощью стандартных статистических тестов. Вариабельность между циклами можно определить путем включения препарата сравнения, значение активности (potency) которого должно укладываться в  0,5 lg от среднего значения, установленного в лаборатории, чтобы методика была приемлемой. При должном обосновании допускается использовать методики с худшей прецизионностью.
0,5 lg от среднего значения, установленного в лаборатории, чтобы методика была приемлемой. При должном обосновании допускается использовать методики с худшей прецизионностью.
5. По возможности в исследованиях по очистке с использованием "релевантных" и специфичных "модельных" вирусов необходимо рассчитывать границы 95% доверительных интервалов для установленного фактора (коэффициента) снижения вирусной нагрузки. Если границы 95% доверительных интервалов для вирусологических методик изучения исходного материала равны  s, а для вирусологических методик изучения материала на следующем этапе обработки равны
s, а для вирусологических методик изучения материала на следующем этапе обработки равны  a, то границы 95% доверительных интервалов для фактора (коэффициента) снижения равны:
a, то границы 95% доверительных интервалов для фактора (коэффициента) снижения равны:
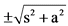 (1)
(1)
Вероятность обнаружения вирусов в низкой концентрации
5. Очевидно, что при низкой концентрации вируса (например, 10 - 1000 инфекционных частиц на литр) проба объемом несколько миллилитров может не содержать инфекционных частиц. Вероятность (p), что такая проба не содержит инфекционных вирусов, равна:
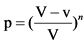 ,
,
где:
V - общий объем подлежащего испытанию материала (л);
v - объем пробы (л);
n - абсолютное количество инфекционных частиц, статистически распределенных в объеме V.
Если V >> v, то уравнение приближенно описывается распределением Пуассона:
p = e-cv,
где c - концентрация инфекционных частиц на литр, тогда величина c определяется по формуле:
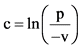
Пример: Если испытуемая проба имеет объем 1 мл, вероятность (p), что в ней не содержится вирус, истинные концентрации которого находятся в диапазоне 10 - 1000 инфекционных частиц на литр, равна:
|
c
|
10
|
100
|
1000
|
|
p
|
0,99
|
0,90
|
0,37
|
То есть при концентрации 1000 вирусов на литр, 37% проб объемом 1 миллилитр не будут содержать вирусных частиц.
6. Если лишь часть проб испытывается на вирусы и результаты отрицательны, необходимо рассчитать количество вирусов, которое должно содержаться в общем количестве проб, чтобы результаты оказались положительными. Полученное значение необходимо учитывать при расчете фактора снижения вирусной нагрузки. Рекомендуется рассчитать 95% доверительные интервалы. Однако в некоторых случаях, ввиду недостаточного количества материала, их расчет невозможен.
Приложение N 4
к главе 2 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
УКАЗАНИЯ
ПО РАСЧЕТУ ФАКТОРОВ (КОЭФФИЦИЕНТОВ) СНИЖЕНИЯ ВИРУСНОЙ
НАГРУЗКИ В ИССЛЕДОВАНИЯХ ДЛЯ ОПРЕДЕЛЕНИЯ ОЧИСТКИ ОТ ВИРУСОВ
Фактор (коэффициент) снижения вирусной нагрузки на отдельном этапе очистки или инактивации выражается в виде десятичного логарифма отношения вирусной нагрузки в материале до очистки к вирусной нагрузке в материале после очистки, готовом для передачи его на следующий этап. Если исходный материал имеет объем v', титр 10a', то вирусная нагрузка равна (v') x (10a'), а продукт, прошедший этап очистки (инактивации) имеет объем v", титр 10a" и его вирусная нагрузка (v") x (10a"), тогда индивидуальные факторы (коэффициенты) снижения (Ri) рассчитываются по следующей формуле:
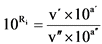
Формула учитывает как титры, так и объемы материалов до и после этапа очистки.
Ввиду неизбежной погрешности при определении титра некоторых вирусов индивидуальный фактор (коэффициент) снижения, используемый для расчета совокупного фактора (коэффициента) снижения вирусной нагрузки, должен превышать 1.
Совокупный фактор (коэффициент) снижения вирусной нагрузки всего процесса производства равен сумме логарифмов индивидуальных факторов (коэффициентов) снижения каждого этапа. Он представляет собой логарифм отношения вирусной нагрузки до начала первого этапа очистки и вирусной нагрузки по завершении всех этапов очистки. Факторы (коэффициенты) снижения, как правило, выражаются в логарифмических координатах, то есть, несмотря на то, что остаточная инфицирующая способность вируса никогда не будет снижена до нуля с математической точки зрения, ее можно значительно снизить на практике.
Приложение N 5
к главе 2 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
УКАЗАНИЯ
ПО РАСЧЕТУ ОЖИДАЕМОГО КОЛИЧЕСТВА ВИРУСНЫХ ЧАСТИЦ НА ДОЗУ
Расчет применим к вирусам, для которых можно рассчитать исходное их количество, например, для эндогенных вирусов.
Пример.
1. Условия. Измеренная или расчетная концентрация вирусов в сборе клеточной культуры равна 106/мл.
Расчетный фактор очистки от вирусов составляет > 1015.
Объем сбора культуры, необходимый для приготовления одной дозы препарата равен 1 л (или 103 мл).
2. Расчет примерного количества частиц на дозу:
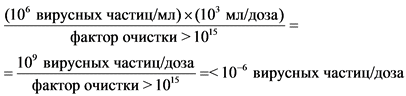
Таким образом, ожидаемое содержание равно менее чем одна частица на миллион доз.
Глава 3. Оценка вирусной безопасности исследуемых
лекарственных препаратов (препаратов для клинических
исследований), полученных с помощью
биотехнологических методов
1. Введение
Обеспечение вирусной безопасности лекарственных препаратов, полученных с помощью биотехнологических методов, является комплексным процессом, при этом надежная оценка вирусной безопасности исследуемого лекарственного препарата (ИЛП) имеет критическое значение. Настоящая глава содержит рекомендации по данным и документации о вирусной безопасности, которые необходимо включать в состав досье на получение разрешения для проведения клинического исследования биотехнологических препаратов для медицинского применения. Следует также руководствоваться главой 2 настоящих Правил, в которой приведены требования к составлению соответствующих разделов регистрационного досье. Несмотря на то, что глава 2 настоящих Правил не содержит рекомендаций в отношении биотехнологических лекарственных препаратов, изучаемых в клинических исследованиях, их основные принципы совпадают и подлежат выполнению.
Настоящая глава содержит гармонизированный подход к оценке вирусной безопасности исследуемых биотехнологических лекарственных препаратов во время клинических исследований как для спонсоров, так и для регулирующих органов. Это особенно ценно при проведении многоцентровых исследований, потенциально охватывающих различные государства-члены.
2. Область применения
Область применения настоящей главы распространяется на исследуемые биотехнологические лекарственные препараты для медицинского применения, произведенные из культивированных in vitro клеток, полученных из охарактеризованных банков клеток человеческого или животного происхождения, указанных в главе 2 настоящих Правил. Многие исследуемые лекарственные препараты получены из хорошо охарактеризованных клеточных линий грызунов, например, CHO, NS0 или SP2/0, хотя также используется и находится в стадии разработки ряд других клеточных линий, использование которых следует рассматривать в индивидуальном порядке.
Положения настоящей главы распространяются на моноклональные антитела и исследуемые лекарственные препараты, полученные методом рекомбинантной ДНК, включая рекомбинантные субъединичные вакцины. Однако требования настоящей главы не применяются в отношении исследуемых лекарственных препаратов, содержащих рекомбинантные вирусы или бактерии (как реплицирующиеся, так и нереплицирующиеся), или живых аттенуированных и инактивированных вакцинах. Исследуемые лекарственные препараты, полученные из гибридомных клеток, выращенных in vivo, также не входят в сферу применения настоящей главы.
В настоящей главе изложены требования к вирусной безопасности, применимые ко всем этапам клинической разработки лекарственных препаратов. Она не распространяется на лекарственные препараты, исследуемые исключительно в доклинических испытаниях. Требования к данным, подлежащим включению в регистрационное досье, содержатся в главе 2 настоящих Правил.
3. Правовая основа
Проведение клинических исследований в государствах-членах регулируется международными договорами и актами, составляющими право Союза, и законодательством государств-членов. Исследуемые лекарственные препараты, изучаемые в клинических исследованиях, должны быть произведены в соответствии с правилами надлежащей производственной практики Союза, утверждаемыми Комиссией.
4. Правила
4.1. Общие принципы
Целью исследований вирусной безопасности биотехнологических исследуемых лекарственных препаратов является подтверждение приемлемого уровня безопасности для субъектов клинических исследований.
Вирусная безопасность зарегистрированного биотехнологического лекарственного препарата обеспечивается тремя взаимодополняющими подходами, включающими в себя:
отбор и испытание клеточных линий и другого сырья человеческого или животного происхождения на вирусную контаминацию;
оценку возможностей последующих после культивирования процессов обработки обеспечивать очистку от инфекционных вирусов;
испытание препарата на определенных этапах производства на наличие контаминирующих вирусов в соответствии с главой 2 настоящих Правил).
В связи с экспериментальным характером производственного процесса и препарата в отношении исследуемых биотехнологических лекарственных препаратов предусматривается сокращенная программа испытаний по обеспечению вирусной безопасности в сравнении с требованиями к данным, необходимым для регистрации лекарственного препарата: во-первых, по объему оценки на наличие вирусов в клетках-продуцентах по завершении производства или в необработанном нерасфасованном продукте в соответствии с подразделом 4.2.3 настоящей главы и, во-вторых, по объему исследований по валидации снижения вирусной нагрузки в соответствии с подразделом 4.2.4 настоящей главы. Такая сокращенная программа будет применима только к клеточным линиям, относимым согласно главе 2 настоящих Правил, к "ситуации 1" и "ситуации 2". Собственный опыт фармацевтического производителя в соответствии с подразделом 4.2.4 настоящей главы также может способствовать сокращению объема исследований вирусной безопасности. Помимо представления данных, оценку риска следует осуществлять с учетом нескольких или всех следующих факторов:
природа и история клеточной линии;
степень установления свойств клеточной линии;
использование сырья человеческого и (или) животного происхождения во время производства и контроля их качества;
возможность контаминации продукта посторонними агентами;
опыт работы производителя с используемой клеточной линией;
опыт применения производителем специальных методик снижения вирусной нагрузки, которые будут использоваться;
опубликованные данные.
4.2. Обеспечение вирусной безопасности биотехнологических
исследуемых лекарственных препаратов
4.2.1. Квалификация клеточной линии: испытание на наличие вирусов.
Перед началом исследований I фазы необходимо выполнить испытание ГБК на наличие вирусной контаминации в соответствии с главой 2 настоящих Правил. Создание РБК возможно только в процессе клинического исследования, поэтому в отношении некоторых исследуемых биотехнологических лекарственных препаратов, используемых на ранних этапах клинического исследования, он может быть еще не создан. После создания первого РБК он должен быть испытан в соответствии с главой 2 настоящих Правил. Тем не менее при испытании необработанного нерасфасованного продукта в соответствии с подразделом 4.2.3 настоящей главы испытание клеток с предельным для производства клеточным возрастом in vitro не требуется.
Поскольку эндогенные ретровирусы или ретровирусоподобные частицы присутствуют в большинстве клеточных линий, используемых в настоящее время, и есть вероятность того, что они будут содержаться в новой клеточной линии, особое внимание следует уделять испытанию клеточной линии на их наличие.
4.2.2. Сырье биологического происхождения.
При оценке вирусной безопасности исследуемых биотехнологических лекарственных препаратов необходимо учитывать биологическое сырье (особенно животного или человеческого происхождения), используемое в производстве. Приемлемыми подходами к оценке его вирусной безопасности являются оценка с учетом рисков вида и происхождения сырья, условий его переработки и испытания, а также его использование в производстве лекарственных препаратов и испытания, проведенные с необработанным нерасфасованным материалом, согласно подразделу 4.2.3 настоящей главы.
В отношении вирусной безопасности сырья биологического происхождения необходимо представить соответствующие документы. Следует руководствоваться документами по обеспечению безопасности при использовании бычьей сыворотки и требованиям Фармакопеи Союза по минимизации риска передачи губчатой энцефалопатии животных.
4.2.3. Испытание необработанного нерасфасованного продукта на наличие вирусов.
Независимо от этапа исследования каждую серию необработанного нерасфасованного продукта, которая будет использоваться для производства материалов для клинического исследования (исследуемого лекарственного препарата), необходимо испытать в соответствии с требованиями главы 2 настоящих Правил. Испытуемый образец должен включать в себя клетки (при необходимости), а испытания - тесты in vitro, ПЦР-скрининг на наличие посторонних агентов и оценку наличия ретровирусоподобных частиц, если это применимо. Для нерасфасованных материалов, полученных из клеточных линий CHO, дополнительных испытаний не требуется. Если производство основано на клеточных линиях NS0 или Sp2/0, испытания на инфекционные ретровирусы следует проводить однократно, но в случае существенных изменений в культурах клеток-продуцентов, например, при переводе производства на промышленный масштаб, их необходимо провести повторно. Если производство основано на иных клеточных линиях, испытания на инфекционные ретровирусы и испытания in vivo, предусмотренные подразделом 3.2.3 главы 2 настоящих Правил, следует проводить однократно, но в случае существенных изменений в культурах клеток-продуцентов, например, при переводе производства на промышленный масштаб, их необходимо провести повторно. Рекомендации по указанным испытаниям представлены в таблице 1.
Таблица 1
Требования к испытаниям необработанного
нерасфасованного продукта
|
Испытания in vitro
|
Испытания на инфекционные ретровирусы <*>
|
Испытания in vivo <*>
|
|
|
CHO
|
Да, все полученные нерасфасованные продукты <**>
|
Нет
|
Нет
|
|
NS0 Sp2/0
|
Да, все полученные балк-препараты <**>
|
Да, 1 раз для заданного масштаба производства
|
Нет
|
|
все иные клеточные линии
|
Да, все полученные балк-препараты <**>
|
Да, 1 раз для заданного масштаба производства
|
Да, 1 раз для заданного масштаба производства
|
--------------------------------
<*> По возможности, для выявления вирусов, ассоциированных с клетками, испытуемый материал должен содержать клетки или фрагменты клеток. В отношении клеточных культур, подвергающихся перфузии, производитель должен определить и обосновать наиболее подходящий этап для отбора проб, содержащих клетки для целей испытаний. Также допускается получение испытуемого материала из клеток, культивированных сверх срока, используемого для производства серии продукта; в этом случае необходимо обосновать выбранный подход. Испытания на инфекционные ретровирусы могут быть опущены, если более чувствительные тесты показали отрицательные результаты.
<**> Количественное определение ретровирусов или ретровирусоподобных частиц следует проводить только в отношении первых трех серий нерасфасованного продукта на определенном этапе разработки (или на меньшем количестве, если произведено менее трех серий нерасфасованного продукта).
Целесообразно включить испытание на мелкий вирус мышей (MMV), если клеточная линия восприимчива к этому вирусу.
При разработке испытаний для необработанного нерасфасованного материала необходимо принять во внимание источник и вирусную безопасность сырья, используемого во время культивирования клеток, согласно подразделу 4.2.2 настоящей главы. Если используется сырье человеческого или животного происхождения, например, бычья сыворотка, могут потребоваться дополнительные специальные испытания.
4.2.4. Валидация методов обеспечения вирусной безопасности.
Целями валидации методов являются характеристика и оценка этапов процесса, которые могут считаться эффективными для инактивации (элиминации) вирусов и количественная оценка общей величины снижения содержания вирусов (вирусных частиц), например эндогенных ретровирусных частиц. В каждом случае потребуется индивидуальный подход с учетом характеристик клеточной линии, использования сырья биологического происхождения, а также природы этапов процесса, которые могут быть эффективными в инактивации (элиминации) вирусов.
Независимо от объема прямых испытаний линии клеток-продуцентов на вирусы, в связи с ограничениями методик обнаружения вирусов сохраняется возможность неизвестной контаминации клеток вирусом, изначально присутствующим в клетках или содержащимся в материале биологического происхождения, используемом во время культивирования клеток-продуцентов. Следовательно, даже если сырье биологического происхождения не используется и клеточная линия полностью проверена, необходимо оценить последующие этапы обработки всех исследуемых лекарственных препаратов на предмет способности инактивировать (элиминировать) вирусы.
Валидацию методов обеспечения вирусной безопасности следует провести до начала клинического исследования. Потенциальными контаминантами могут быть оболочечные и безоболочечные вирусы, и исследования по вирусной безопасности должны включать определение как оболочечных, так и малых безоболочечных вирусов, желательно парвовирусов. Необходимо подтвердить, что все вирусы или вирусные частицы, о наличии которых в нерасфасованном сборе было заведомо известно, эффективно инактивированы или элиминированы в ходе последующей обработки продукта. В ситуации 2, предусмотренной главой 2 настоящих Правил, в случае содержания эндогенных ретровирусов или ретровирусоподобных частиц, при валидации инактивации (элиминации) вирусов необходимо использовать ретровирус для подтверждения полной очистки от частиц, присутствующих в нерасфасованном сборе.
Исследования по снижению вирусной нагрузки следует осуществлять в соответствии с принципами, указанными в главе 2 настоящих Правил, однако не всегда требуется подтверждение устойчивости (то есть влияние показателей процесса производства на снижение вирусной нагрузки). Необходимо представить описание соответствующих этапов очистки препарата, способствующих обеспечению вирусной безопасности. Во время инактивации (элиминации) потенциальных вирусов на этих этапах следует учитывать вирусную безопасность линии клеток-продуцентов, например, вид и степень эндогенной ретровирусной контаминации или использование во время производства материалов человеческого или животного происхождения, а также возможную степень контаминации. Глава 4 настоящих Правил также содержит подробную информацию об этих исследованиях.
Желательно исследовать влияние более чем 1 этапа производства на обеспечение вирусной безопасности и произвести оценку не менее 2 ортогональных стадий. Ортогональные стадии представляют собой стадии процесса, характеризующиеся разными механизмами инактивации (элиминации) вирусов. Критерии эффективности приведены в главе 4 настоящих Правил. Не требуется исследовать этапы производства, которые не предполагают существенного обеспечения вирусной безопасности.
Воспроизводимость эффективного этапа обеспечения вирусной безопасности должна быть подтверждена не менее чем в 2 независимых экспериментах.
При проведении валидационного исследования необходимо использовать предельные параметры технологического процесса (наихудший сценарий), если они известны. Однако в ходе разработки такие наихудшие предельные параметры нового технологического процесса могут быть не установлены. В этих случаях обоснованно применение репрезентативных (модельных) условий при подтверждении производителем осуществления фактического процесса производства в заданном режиме.
Сокращению объема указанных исследований способствуют следующие условия:
исследование одного конкретного этапа инактивации (элиминации) вирусов может быть достаточным, если во время этого этапа может быть продемонстрировано эффективное снижение вирусной нагрузки, обусловленной широким спектром вирусов, включая такие малые безоболочечные вирусы, как парвовирусы. Однако для подтверждения полноценной очистки клеток, подпадающих под ситуацию 2, от ретровирусных частиц обычно необходимо провести оценку более чем 1 этапа производства;
необходимо учитывать предыдущий опыт производителя по использованию определенных стадий производства, следующих после культивирования. Если производитель разрабатывает подобные типы препаратов путем применения отработанных и хорошо охарактеризованных процессов производства, данные по снижению вирусной нагрузки таких препаратов могут быть применены к новому препарату на эквивалентном этапе производства.
Как правило, чтобы использовать данные по этому этапу производства, он должен пройти тщательную проверку, включая детальное изучение параметров процесса производства, обеспечивающих снижение вирусной нагрузки. Если в отношении конкретного этапа есть данные, которые можно использовать для более чем одного препарата, эффективность обеспечения вирусной безопасности в каждом случае должна быть сопоставимой. Обработка нового и уже производимого ранее препарата (препаратов) до этого конкретного этапа должна осуществляться по сходной стратегии.
Необходимо представить обоснование применения внутренних данных разработчика для производства нового препарата, например, на данные об обеспечении вирусной безопасности на определенном этапе производства можно будет ссылаться, если промежуточный продукт, полученный на стадии, предшествующей этому этапу, имеет сопоставимые биохимические свойства и проходит очистку идентичными методами. Производитель должен представить результаты критического анализа этапа производства, на котором будут применяться внутренние данные разработчика, и состав соответствующего промежуточного продукта. Эти сведения должны включать в себя, например, тип фильтра, удельную нагрузку на фильтр, скорость потока, давление и состав промежуточных продуктов (в отношении вирусных фильтров) или размеры колонок, включая высоту слоя наполнителя, нагрузку, состав буферного раствора и промежуточных продуктов производства, а также линейные скорости потоков для хроматографических методов. Помимо этого, каждый новый препарат может иметь компоненты, не содержащиеся в предыдущих препаратах, поэтому необходимо учитывать потенциальное влияние компонентов, специфичных для этого препарата. Анализ должен полностью подтвердить вывод о том, что в обоих случаях используемый этап производства имеет аналогичную способность инактивировать (элиминировать) потенциальные вирусные контаминанты. Если сравнение этого этапа неубедительно или база данных недостаточно убедительна, чтобы исключить опосредованное препаратом влияние на способность процесса снижать вирусную нагрузку, для подтверждения фактической эффективности этапа необходимо осуществить не менее одного цикла обработки с соответствующим вирусом. Если показатели технологического процесса явно отличаются, например, при использовании аналогичного оборудования получены различные хроматографические профили, этап подлежит валидации в соответствии с указанной методикой и принципами, предусмотренными в главе 2 настоящих Правил.
Опубликованные данные могут оказаться полезными для определения возможностей этапа инактивировать (элиминировать) вирусы, а также способствовать пониманию механизмов, лежащих в их основе. Это упрощает изучение ключевых технологических параметров, влияющих на снижение вирусной нагрузки, и установление наихудших предельных значений на определенных этапах, подлежащих валидации. Тем не менее применение опубликованных факторов снижения вирусной нагрузки к конкретному препарату потребует расширенного подтверждения сопоставимости технологических процессов, промежуточных продуктов и гарантирования того, что опосредованные препаратом факторы производства не влияют на снижение вирусной нагрузки. Снижение вирусной нагрузки может зависеть от разных технологических параметров и конкретного состава промежуточного продукта. Более того, заданная способность по снижению вирусной нагрузки может быть специфична для выбранных вирусов (например, при использовании хроматографических методов). Именно поэтому опубликованные данные должны быть тщательно проанализированы.
В связи с использованием специализированных колонок и малым количеством серий препарата на ранних этапах разработки проведение в отношении исследуемых лекарственных препаратов специальных исследований по повторному использованию и регенерации колонок, как правило, не требуется. Тем не менее во всех случаях интенсивного повторного использования колонок для производства исследуемых лекарственных препаратов этот факт следует учитывать при исследовании способности процесса снижать вирусную нагрузку.
Ревалидация методов снижения вирусной нагрузки.
Данные об исследуемых лекарственных препаратах, использованных в предыдущем исследовании (например, первом исследовании I фазы), могут быть применены в последующих исследованиях. Однако во время разработки исследуемых лекарственных препаратов могут произойти значительные изменения в процессе производства, и эти изменения могут повлиять прямо или косвенно (если изменения не учитывались во время оценки этапов производства) на способность снижения вирусной нагрузки. Следовательно, если имеющиеся данные не отражают производство исследуемых лекарственных препаратов, используемых в предстоящем клиническом исследовании, повторная оценка должна быть произведена до начала следующих клинических исследований. В зависимости от введенных изменений следует пересмотреть подбор вирусов и при необходимости ввести дополнительные вирусы для обеспечения возможностей процесса снижать вирусную нагрузку. Даже если в соответствии с главой 2 настоящих Правил в продленных клинических исследованиях на поздних стадиях (например, III фазы) полная валидация не требуется, производители должны обосновать выбранный подход с учетом "модельных" вирусов и оценки этапов процесса производства. В соответствии с главой 2 настоящих Правил полномасштабные валидационные исследования вирусной безопасности необходимо проводить после создания окончательного процесса производства и очистки.
4.2.5. Описание и аттестация аналитических методик.
Для испытания исходного материала и промежуточных продуктов на наличие вирусов или при оценке способности процесса снижать вирусную нагрузку могут использоваться различные аналитические методики. Методы исследований по обнаружению вирусов включают в себя широкий спектр испытаний in vitro по оценке цитопатического действия (ЦПД) и гемоадсорбции на множестве индикаторных клеточных линий, испытаний in vivo и специфических испытаний на наличие вирусов с применением, например, ПЦР. Для проведения испытаний на наличие ретровирусов могут быть использованы трансмиссионная электронная микроскопия (ТЭМ), анализ методом совместного культивирования с использованием разных линий клеток и исследования на обратную транскриптазу (например, усиленная в препарате обратная транскриптаза (PERT)).
Независимо от фазы клинического исследования необходимо обосновать пригодность аналитических методик, используемых для проверки на наличие вирусов (как качественных, так и количественных методов). Как правило, применяются подразделы 3.2 и 4 главы 2 настоящих Правил. Чтобы дать точное представление об используемом методе и способе его контроля, следует представить достаточно подробное описание аналитических методик, включая реагенты, контроли, процедуру испытания и критерии валидности. При использовании фармакопейных методик необходимо привести точные ссылки.
При необходимости в отношении аналитических методик, использованных при квалификации системы банков клеток и других исходных материалов, так же, как для испытания необработанного нерасфасованного материала на наличие вирусов, следует представить резюме результатов квалификации и (или) валидации аналитических методик в формате таблицы (например, результаты значений, полученных в отношении специфичности с применением положительного и отрицательного контроля, чувствительности, количественного определения и предела обнаружения). Полный отчет о квалификации каждого метода представлять не следует, но необходимо иметь их в наличии для представления по запросу уполномоченных органов государств-членов.
В отношении дополнительных аналитических методик, используемых в исследованиях по снижению вирусной нагрузки, необходимо представить подробную информацию, подтверждающую пригодность этих методик для определения количества вирусных частиц. Информация должна включать в себя описание исследований по оценке, например, предела количественного определения, специфичности, вариабельности внутри исследования, влияния буферного раствора (матрицы) на определение вирусной инфекционности, а также цитотоксичности препарата и буферного раствора, которые могут повлиять на способность выбранных "модельных" вирусов инфицировать индикаторные клетки. Рекомендации по статистической оценке результатов вирусологических испытаний приведены в приложении N 3 к главе 2 настоящих Правил. Если применимо, возможно принятие отчета контрактной лаборатории, выполнившей проверку на наличие вирусов.
4.3. Оценка риска вирусной безопасности
Помимо представления данных о вирусной безопасности препарата, вместе с заявлением о получении разрешения на проведение клинического исследования необходимо представить оценку риска вирусной безопасности. Основными факторами следует считать факторы, указанные в подразделах 4.1 и 4.2.1 - 4.2.4 настоящей главы. В соответствии с главой 2 настоящих Правил необходимо учитывать результаты испытаний клеточной линии и всего сырья человеческого и животного происхождения на наличие вирусных контаминантов, валидацию вирусной безопасности и проверку препарата на соответствующих этапах процесса производства на отсутствие инфекционных вирусов.
Оценка риска должна включать в себя определение количества предполагаемых частиц на дозу в соответствии с приложением N 5 к главе 2 настоящих Правил и содержать все этапы производственного процесса.
В отдельных случаях при оценке совокупного риска для клинического исследования целесообразно рассмотреть такие клинические параметры, как показание к применению, доза, частота применения, число лиц, которые подвергнутся экспозиции, длительность исследования и иммунологический статус пациентов. В этом контексте следует учесть, что некоторые из этих параметров могут измениться в период между фазами I, II и III. Клинические параметры не должны рассматриваться в качестве параметров, используемых для принятия первоочередных решений, но они могут учитываться при принятии заключительного решения о выдаче разрешения на проведение клинического исследования с точки зрения вирусной безопасности.
Каждую ситуацию следует рассматривать индивидуально.
4.4. Повторная оценка вирусной безопасности
во время исследования
Во время испытаний исследуемых лекарственных препаратов часто вносятся технологические изменения, при этом некоторые из них могут повлиять на ранее определенную оценку вирусной безопасности. Во всех случаях внесения изменений в процесс производства исследуемого лекарственного препарата, для которого уже была выполнена оценка риска вирусной безопасности, производитель должен документировать все внесенные изменения и принять решение о необходимости переоценки риска по каждому из них. В некоторых случаях будет ясно, что изменение не влияет на оценку риска вирусной безопасности. Однако при явном влиянии или неопределенности исхода следует провести повторную оценку риска и в необходимых случаях провести надлежащие экспериментальные исследования. В этих случаях необходимо учитывать все аспекты обеспечения вирусной безопасности.
В отношении изменений, которые могут снизить валидность исследований по вирусной безопасности, следует обратиться к подразделу "Ревалидация методов снижения вирусной нагрузки" подраздела 4.2.4 настоящей главы.
4.5. Формат документации для получения разрешения
на проведение клинического исследования
В досье для получения разрешения на проведение клинического исследования необходимо включить раздел, содержащий документы, касающиеся вирусной и ТГЭ безопасности. Все данные должны быть собраны воедино с минимальным количеством ссылок на другие разделы основного досье, что позволит выполнить их единую оценку. Полные отчеты, включая исходные данные об испытании клеточных линий и исследования по вирусной безопасности, необходимо представлять по требованию. Во время оценки представленных данных уполномоченные органы государств-членов вправе запросить эти отчеты, чтобы получить наиболее точное и ясное представление о вирусной безопасности исследуемых лекарственных препаратов. Исходные данные могут быть представлены контрактными или собственными лабораториями как часть отчета. Если заявитель использует ранее полученные собственные данные (то есть данные по другим препаратам), необходимо представить достаточный комплект данных, позволяющих оценить собственные данные и получить уверенность в том, что они валидны или обосновывают разработку исследуемого лекарственного препарата.
Глава 4. Валидация методов обеспечения вирусной
безопасности: разработка, проведение и интерпретация
результатов исследований по валидации методов инактивации
и удаления вирусов
1. Введение
1.1. В настоящей главе рассматривается необходимость проведения и влияния валидационных исследований на вирусы на обеспечение вирусной безопасности биологических препаратов. Основными целями настоящей главы являются представление рекомендаций по планированию валидационного исследования, включая выбор используемых вирусов, и интерпретация полученных данных, особенно в отношении определения этапа процесса, который можно считать эффективным для инактивации и (или) элиминации вирусов.
1.2. В настоящей главе рассматривается валидация процедур инактивации и (или) элиминации вирусов из всех категорий биологических лекарственных препаратов для медицинского применения, за исключением живых вирусных вакцин (в том числе генно-инженерных живых векторов). Виды рассматриваемых препаратов:
препараты, получаемые при культивировании in vitro клеточных линий человеческого или животного происхождения;
препараты, получаемые при культивировании in vivo или из органов и тканей человека или животных;
препараты, полученные из крови или мочи, или других биологических жидкостей человека или животных.
1.3. Риск вирусной контаминации характерен для всех биологических препаратов, производство которых предполагает использование материала животного или человеческого происхождения. Вирусная контаминация биологического препарата может быть обусловлена исходным материалом, например, банками клеток животного происхождения, кровью человека, тканями человека или животных, посторонние агенты могут быть привнесены в процесс производства, например, при использовании сыворотки животных при культивировании клеток.
1.4. Основной причиной передачи вирусов, предусмотренной пунктом 1.3 настоящей главы, является контаминация исходных материалов. Контаминация биологического препарата может также происходить при использовании инфицированного материала в процессе производства или с вспомогательным веществом. В некоторых случаях вирус может быть обнаружен спустя годы после ввода препарата в обращение, поскольку контаминация произошла до получения достаточных знаний о наличии инфекционных агентов (вакцина для профилактики желтой лихорадки, может быть контаминирована вирусом лейкоза птиц, который естественным образом инфицирует куриные яйца, и вирусом гепатита B, содержащимся в сыворотке человека, использованной в качестве стабилизатора, вирус SV40 может контаминировать вакцины для профилактики полиомиелита и аденовирусной инфекции, производимые на первичных культурах клеток почек, взятых от макак-резусов, которые являются естественным резервуаром вируса SV40). Кроме того, вирусы, содержащиеся в плазме человека, например, ВИЧ и вирус гепатита C, могут контаминировать препараты крови.
1.5. В целях контроля потенциальной вирусной контаминации биологических лекарственных препаратов можно использовать три основных взаимодополняющих подхода:
отбор и испытание исходных материалов на предмет отсутствия обнаруживаемых вирусов;
испытание способности производственных процессов элиминировать или инактивировать вирусы;
испытание препарата на соответствующих этапах производства на предмет отсутствия обнаруживаемых вирусов.
Ни один из подходов сам по себе не дает достаточной гарантии, поэтому в целях ее достижения необходимо использовать их комбинацию.
1.6. Испытание исходных материалов является обязательным условием минимизации вирусной контаминации. Испытания могут обнаруживать один или более видов вирусов, однако ни одно отдельное испытание не способно подтвердить присутствие всех известных вирусов. Более того, в целях получения положительного результата любые аналитические системы требуют некоторой минимальной вирусной контаминации, испытания также ограничены статистическими погрешностями при отборе проб. Некоторые испытания, например, на антитела к вирусному гепатиту C в плазме человека, способны измерять маркеры инфекции, которые появляются спустя некоторое время после инфицирования. Аналогичные соображения справедливы и в отношении испытания лекарственного препарата.
1.7. В связи с этим подтверждение отсутствия в биологическом лекарственном препарате инфекционных вирусов во многих случаях происходит не только за счет прямого испытания на их наличие, но также путем подтверждения того, что процесс производства способен элиминировать или инактивировать их. Валидация процесса инактивации и (или) элиминации вирусов может играть ключевую роль в установлении безопасности биологических препаратов, особенно при высокой вероятности контаминации исходного материала и сырья патогенными для человека вирусами, например, препаратов, полученных из плазмы. Кроме того, поскольку во многих случаях в прошлом происходила контаминация агентами, о которой не было известно, и даже отсутствовали подозрения о такой возможности на момент производства, оценка процесса производства может дать определенную уверенность в том, что широкий спектр вирусов (включая неизвестные опасные вирусы) подвергается элиминации.
1.8. Целью настоящей главы является описание общих принципов проведения валидационных исследований и вирусологического подхода, который необходимо использовать при планировании валидационных исследований на вирусную нагрузку. Производители должны использовать правила, содержащиеся в настоящей главе, в отношении конкретного препарата с учетом свойств исходного материала и сырья, используемых при производстве и в процедурах очистки, а также любых других факторов, которые могут влиять на этот аспект безопасности. Производители должны объяснить и обосновать подход, использованный ими в исследованиях по оценке элиминации вирусов, в регистрационном досье.
2. Источники вирусной контаминации
Вирусная контаминация биологических препаратов может быть обусловлена следующим.
2.1. Исходный материал и сырье могут быть контаминированы вирусами, инфицирующими вид животных, являющийся источником материала или сырья. Кровь может содержать различные вирусы, и применение препаратов, полученных из плазмы человека, приводило к инфицированию вирусами гепатитов B и C, ВИЧ, парвовирусом B19 и иногда вирусом гепатита A. Мышиные вирусы, некоторые из которых патогенны для человека, могут контаминировать мышиные гибридомы. Клеточные линии, предназначенные для осуществления генетической манипуляции, могут быть контаминированы вирусами, в связи с чем необходимо внимательно подходить к их выбору и проводить испытания на предмет отсутствия обнаруживаемых посторонних агентов еще до генетической манипуляции, чтобы начать работу с должным образом охарактеризованной клеточной линией.
2.2. Клетки могут быть источником латентной или персистирующей инфекции, например, вируса герпеса или ретровируса, которые могут передаваться вертикально от одного поколения клеток к следующему в виде вирусного генома и которые могут периодически экспрессироваться в качестве инфекционного вируса.
2.3. При создании производственной клеточной линии может быть привнесен контаминирующий вирус, характерный для других видов животных, например, трансформированная вирусом Эпштейна-Барр лимфобластоидная клеточная линия человека, секретирующая моноклональные антитела, может быть инфицирована мышиным ретровирусом после слияния с клетками миеломы мышей.
2.4. Посторонние вирусы могут быть привнесены при использовании в процессе производства контаминированных животных продуктов, например, клеточные культуры могут быть контаминированы бычьими вирусами вследствие использования бычьей сыворотки, или мышиные моноклональные антитела, используемые в аффинной хроматографии, могут контаминировать препарат вирусами мышей.
2.5. Возможны иные источники контаминации, например, производственный персонал или сырье небиологического происхождения.
3. Процесс валидации
3.1. Цель валидационных исследований по очистке от вирусов заключается в:
подтверждении того, что процесс производства эффективно инактивирует и (или) элиминирует вирусы, которые известны своей способностью контаминировать исходные материалы и сырье, или вирусы, которые, предположительно, обладают такими свойствами;
непрямом подтверждении того, что процесс производства способен инактивировать и (или) элиминировать новые или непредвиденные вирусы.
Это достигается за счет преднамеренного добавления (spiking) вируса к материалу, находящемуся на различных этапах производства, и измерения степени его элиминации или инактивации в ходе последующих этапов. Этот подход позволяет определить этапы производства, которые эффективны в обеспечении снижения содержания инфекционного вируса; он характеризует общую способность процесса элиминировать инфекционность контаминирующих вирусов.
3.2. Валидационные исследования на вирусы, подобно прямому испытанию материалов на соответствующих этапах, влияют на обеспечение вирусологической безопасности препарата. Вместе с тем, все валидационные исследования на вирусы следует рассматривать как некоторую приближенную оценку истинных возможностей процесса, поскольку проведение идеального валидационного исследования процесса производства может оказаться затруднительным в связи с вовлечением большого числа сложных переменных. Результаты свидетельствуют о том, что даже незначительные модификации в процедуре или определенном лабораторном штамме используемого вируса могут оказывать большое влияние на элиминацию и инактивацию вирусов.
3.3. Если исходный материал или сырье недостаточно охарактеризованы, например, кровь, ткани и органы человека или животных, или если культивирование клеток осуществлялось в условиях in vivo, повышается вероятность вирусной контаминации, поэтому процесс производства, как правило, должен включать в себя один или несколько эффективных этапов инактивации и (или) элиминации вирусов. Препараты, полученные из плазмы, вызывают особые опасения в отношении вирусной безопасности.
3.4. Допускается что, если исходный материал (например, полностью охарактеризованный банк клеток) представляет меньший вирусологический риск, процесс очистки, как правило, не включает специальный этап инактивации и (или) элиминации вирусов, и считается, что валидированный процесс очистки обеспечивал достаточную степень инактивации и (или) элиминации вирусов. Имеющийся клинический опыт не обнаружил какие-либо недостатки этого подхода. Тем не менее некоторые производители моноклональных антител (МкАТ) используют специальные этапы инактивации и (или) элиминации вирусов в процессе производства, поскольку клеточные линии-продуценты МкАТ мышиного происхождения выделяют различное количество потенциально инфекционных ретровирусов.
3.5. Следует помнить, что системам культур клеток присуще поддержание репликации вирусов. В связи с этим, несмотря на то, что банк клеток хорошо охарактеризован, сохраняется риск вирусной контаминации культуры, имеются сообщения о единичных случаях контаминации посторонними вирусами.
3.6. Обоснование проведения требуемых валидационных исследований и их объем зависят от процесса производства и типа препарата (например, вида животных, являющихся источником исходного материала, степени вариабельности исходного материала и сырья, стабильности активного продукта и т.д.). Достаточность исследований будет определяться в индивидуальном порядке.
4. Выбор вирусов для валидации
4.1. Вирусы для валидации должны, во-первых, быть как можно ближе по своей природе к вирусам, которые могут контаминировать препарат, а во-вторых, обладать как можно более широким диапазоном физико-химических свойств, чтобы определить способность системы элиминировать вирусы в целом.
4.2. В большинстве валидационных исследований используются штаммы вирусов, которые легко получить и количественно определить. Различные лабораторные штаммы вирусов могут обладать отличными друг от друга, а также от естественно встречающихся вирусов свойствами. Следовательно, любой вирус, использованный в валидационном исследовании, является фактически "модельным" вирусом. Производитель должен обосновать выбор вирусов в соответствии с целями валидационного исследованиями и принципами, указанными в настоящей главе. В случае если два сходных вируса можно использовать для валидационных исследований из-за их большого сходства с возможными контаминантами либо из-за сходства их свойств, следует использовать более резистентный вирус, если не обосновано иное.
4.3. Примеры выбора вирусов
Концентраты факторов свертывания, полученные из плазмы человека, подвергались контаминации ВИЧ. В связи с этим производство подобных материалов необходимо оценить на способность процесса очистки от вирусов инактивировать и (или) элиминировать инфекционный ВИЧ.
Клеточные линии, полученные от грызунов, как правило, содержат эндогенные ретровирусные частицы, которые могут быть инфекционными (частицы C-типа) или неинфекционными (частицы A-типа). Если исходный материал или сырье получают из клеточных линий грызунов, процесс производства необходимо оценить на его способность инактивировать и (или) элиминировать один из близкородственных лабораторных ретровирусов мышей.
Примерами вирусов, обладающих широким диапазоном физико-химических свойств, использованных для оценки общей способности процесса элиминировать вирусную инфекционность, являются:
SV40, полиовирус и парвовирус животных - в качестве небольших безоболочечных вирусов;
парагрипп или мышиный ретровирус - в качестве крупных оболочечных РНК-вирусов;
герпесвирус - в качестве крупного ДНК-вируса.
Примеры вирусов, использованных в прошедших валидационных исследованиях, приведены в таблице.
Таблица
Примеры вирусов,
использованных в валидационных исследованиях на вирусы
|
Вирус
|
Семейство
|
Род
|
Естественный хозяин
|
Геном
|
Оболочка
|
Размер (нм)
|
Форма
|
Резистентность к физико-химической обработке
|
|
Вирус везикулярного стоматита
|
рабдовирусы
|
везикуловирус
|
лошадь, корова
|
РНК
|
да
|
70 x 150
|
пуля
|
низкая
|
|
Вирус парагриппа
|
парамиксовирусы
|
парамиксовирус
|
различные
|
РНК
|
да
|
100 - 200
|
плеосфера
|
низкая
|
|
Вирус иммунодефицита человека
|
ретровирусы
|
лентивирус
|
человек
|
РНК
|
да
|
80 - 100
|
сферическая
|
низкая
|
|
Вирус лейкоза мышей
|
ретровирусы
|
онковирус c типа
|
мышь
|
РНК
|
да
|
80 - 110
|
сферическая
|
низкая
|
|
Вирус Синдбис
|
тогавирусы
|
альфавирус
|
человек?
|
РНК
|
да
|
60 - 70
|
сферическая
|
низкая
|
|
Вирус бычьей вирусной диареи
|
тогавирусы
|
пестивирус
|
корова
|
РНК
|
да
|
50 - 70
|
плеосфера
|
низкая
|
|
Вирус псевдобешенства
|
герпесвирусы
|
варицелловирус
|
свинья
|
ДНК
|
да
|
120 - 200
|
сферическая
|
средняя
|
|
Вирус полиомиелита 1 типа
|
пикорнавирусы
|
энтеровирус
|
человек
|
РНК
|
нет
|
25 - 30
|
икосаэдрическая
|
средняя
|
|
Вирус энцефаломиокардита
|
пикорнавирусы
|
кардиовирус
|
мышь
|
РНК
|
нет
|
25 - 30
|
икосаэдрическая
|
средняя
|
|
Реовирус 3
|
реовирусы
|
ортореовирус
|
различные
|
РНК
|
нет
|
60 - 80
|
сферическая
|
средняя
|
|
Гепатит A
|
пикорнавирусы
|
гепатовирус
|
человек
|
РНК
|
нет
|
25 - 30
|
икосаэдрическая
|
высокая
|
|
SV40
|
паповавирусы
|
полиомавирус
|
обезьяна
|
ДНК
|
нет
|
40 - 50
|
икосаэдрическая
|
очень высокая
|
|
Парвовирусы (собак, свиней)
|
парвовирусы
|
парвовирус
|
собака, свинья
|
ДНК
|
нет
|
18 - 24
|
икосаэдрическая
|
очень высокая
|
Данная таблица содержит неполный перечень вирусов, использованных в валидационных исследованиях. Следовательно, необязательно использовать вирусы, указанные в таблице. Производителям рекомендуется использовать и другие вирусы, особенно если они более пригодны для конкретных производственных процессов.
4.4. Необходимо располагать эффективным, чувствительным и надежным методом количественного определения инфекционности использованных вирусов. Предпочтительно использовать вирусы, которые можно получить в высоком титре, однако это не всегда достижимо.
4.5. Препараты, получаемые из овечьих, козьих и бычьих тканей, могут быть контаминированы такими агентами трансмиссивной губчатой энцефалопатии, как скрейпи, которые накапливаются в центральной нервной системе и лимфоидной ткани. Указания по этим агентам приведены в фармакопее Союза, утверждаемой Комиссией, и отдельной главе настоящих Правил.
5. Дизайн валидационных исследований
5.1. Валидационные исследования предполагают намеренное добавление вирусов на различных этапах производства и измерение степени его элиминации (инактивации) в ходе последующего специального этапа или этапов производства. Необязательно валидировать каждый отдельный этап процесса производства. Предметом валидационного исследования необходимо сделать лишь те этапы, которые, вероятнее всего, вносят вклад в инактивацию (элиминацию) вируса.
5.2. Правила надлежащей производственной практики Союза, утверждаемые Комиссией, не допускают намеренное привнесение какого-либо вируса в производственные технологические линии. В связи с этим валидацию необходимо проводить на отдельном лабораторном оборудовании, предназначенном для вирусологической работы, на разукрупненной (уменьшенной) версии технологической линии, ей должен заниматься персонал, обладающий опытом работы по вирусологии и промышленной биоинженерии. Исследования необходимо проводить в соответствии с правилами надлежащей лабораторной практики Союза, утверждаемыми Комиссией.
5.3. В целях оценки вирусной безопасности препарата обязательным условием приемки результатов, полученных на разукрупненной системе, является сопоставимость модельных и полномасштабных процедур. В связи с этим необходимо путем сравнения таких параметров процесса, как pH, температура, концентрация белка и других компонентов, время реакции, высота колонки (column bed height), линейная скорость потока, отношение скорости потока к высоте (bed height), профиль элюирования и эффективность этапа (например, выход, баланс, специфическая активность, состав), подтвердить валидность разукрупнения. Необходимо проанализировать неизбежные отклонения с точки зрения их потенциального влияния на результаты.
5.4. По возможности необходимо показать, за счет какого процесса достигается снижение вирусной инфекционности (инактивации вируса или элиминации вирусных частиц). Этого можно достичь посредством определения кинетики снижения и (или) баланса вирусной нагрузки, исходя из обстоятельств. Процесс, снижающий вирусную инфекционность, потенциально легче поддается моделированию, чем процессы, обеспечивающие элиминацию частиц. Необходимо изучить кинетику инактивации этапа инактивации вирусов и описать ее в отчетах в табличном и графическом формате. Если слишком быстрая инактивация не позволяет отразить кинетику вирусной нагрузки с помощью описания и регистрации условий процесса, необходимо провести дополнительные исследования, чтобы доказать, что инфекционность действительно снижается за счет инактивации. Таким образом, необходимо внедрить надлежащие контроли, направленные на обнаружение возможного искажающего влияния образца или матрицы, в которую привнесен вирус, на методику количественного определения с установлением пределов обнаружения.
5.5. Необходимо изучить производственные параметры, влияющие на эффективность инактивации (элиминации) вирусов с помощью данного этапа производства, и привести результаты, использованные для установления надлежащих внутрипроизводственных пределов содержания вирусной нагрузки. К критическим параметрам относятся:
такие механические параметры, как скорость потока, скорость перемешивания, размеры колонок, повторное использование колонок и т.д.;
такие физико-химические параметры, как содержание белка, pH, температура, содержание влаги и т.д.
5.6. Антитела, содержащиеся в исходном материале, могут влиять на поведение вируса на этапах разделения и инактивации. В валидационных исследованиях необходимо учесть данное обстоятельство.
5.7. Валидность достигаемого лог-снижения вирусной нагрузки определяется влиянием вариации критических параметров процесса, использованных для установления внутрипроизводственных пределов.
5.8. Опубликованные работы по способности схожих или таких же процессов инактивировать (элиминировать) вирусы могут позволить выявить наиболее эффективные этапы. Однако присущая валидационным исследованиям и обусловленная необходимостью моделирования процесса, выбора используемых вирусов и определения параметров полномасштабного производства в лабораторном масштабе вариабельность означает, что данные о валидации должны основываться на экспериментальных исследованиях, представленных самим заявителем.
5.9. Количество добавляемого в исходный материал вируса на производственном этапе, подлежащем изучению, должно быть как можно большим, чтобы определить способность производственного этапа должным образом инактивировать (элиминировать) вирусы. Однако количество добавляемого вируса не должно существенно нарушать состав производимого материала (количество добавляемого вируса обычно составляет менее 10%). По возможности вычисленные факторы снижения вирусной нагрузки должны основываться на количестве вируса, которое можно обнаружить в исходном материале после добавления к нему вируса, а не на количестве исходного добавленного в материал вируса.
5.10. По возможности вирус из образцов, взятых из модельных экспериментов, следует титровать без дальнейших манипуляций, таких как ультрафильтрация. Если дальнейшая обработка неизбежна (например, удаление ингибиторов или токсических веществ) или предполагается хранение, направленное на обеспечение одновременного титрования всех образцов, необходимо включить надлежащие контроли с целью определения, какое влияние оказывают эти процедуры на результат исследования. Влияние образца на систему обнаружения, включая токсические эффекты, необходимо документировать, поскольку образцы влияют на пределы обнаружения.
5.11. Количественное определение инфекционности необходимо проводить в соответствии с принципами, определенными правилами надлежащей лабораторной практики Союза, утверждаемыми Комиссией, они могут включать бляшкообразование, обнаружение других цитопатических эффектов, таких как образование синцития или очагов, титрование по конечным точкам (например, методики определения TCID50), обнаружение синтеза вирусных антигенов и другие методы. С целью обеспечения надлежащей статистической правильности результата метод должен обладать достаточной чувствительностью и воспроизводимостью и проводиться с достаточным количеством повторов и контролями в соответствии с приложением N 1 к настоящей главе.
5.12. Методы амплификации нуклеиновых кислот (например, ПЦР) являются подходом, обладающим большой чувствительностью для выявления вирусных геномов, они способны обнаруживать такие вирусы, как гепатит B и C, которые не растут на культурах клеток. Однако важным ограничением этой технологии является обнаружение инактивированного вируса в методике амплификации генома, что может занижать степень вирусной инактивации, достигнутой с помощью потенциально эффективного процесса. ПЦР обладает высокой чувствительностью в исследованиях процессов, зависящих от элиминации вирусов. Основными затруднениями при использовании этой технологии являются квантификация, стандартизация, контроль качества и интерпретация результатов. Необходимо однозначно валидировать и стандартизировать методики ПЦР перед внедрением их в процесс валидации, необходимо проявлять предельную осторожность при интерпретации как положительных, так и отрицательных результатов.
5.13. Необходимо обеспечить надлежащее уничтожение любого вируса, который потенциально мог задержаться в системе, перед повторным использованием данной системы (например, путем очистки колонок и т.д.).
6. Интерпретация данных
6.1. При определении эффективности этапа инактивации (элиминации) вирусов необходимо учитывать комбинацию факторов. Оценка этапа исключительно на основании количества инактивированного (элиминированного) вируса может стать причиной ошибочного заключения о том, что процесс, обеспечивающий определенную степень снижения содержания вирусов, будет приводить к получению безопасного препарата. Следующие факторы влияют на эффективность этапа инактивации (элиминации) вирусов, поэтому в каждом случае их необходимо тщательно оценивать:
правильность использованных испытуемых вирусов в соответствии с разделом 4 настоящей главы;
дизайн валидационных исследований в соответствии с разделом 5 настоящей главы;
достигнутое значение снижения вирусной нагрузки (lg). Факторы снижения вирусной нагрузки, равные или превышающие 4,0 lg, свидетельствуют об однозначном влиянии на конкретный исследуемый испытуемый вирус. Вместе с тем следует отметить, что численное значение фактора снижения вирусной нагрузки нельзя использовать в качестве единственной, абсолютной меры эффективности этапа;
кинетику инактивации, которая указывает, является ли измеренный лог-фактор снижения консервативной (стабильной) оценкой. Инактивация вирусов, как правило, не является простой реакцией первого порядка, она обычно состоит из быстрой начальной фазы с последующей медленной фазой. Существенное снижение скорости инактивации со временем может свидетельствовать о снижении эффективности инактивирующего агента или о том, что остаточное содержание вирусов устойчиво к инактивирующему агенту, а это значит, что этап не является ни высокоэффективным, ни устойчивым;
характер инактивации (элиминации) и ее селективность в отношении лишь определенных классов вирусов. Этап процесса инактивации (элиминации) вирусов может быть высокоэффективен в отношении одних вирусов и неэффективен в отношении других (например, обработка Р/Д эффективна против оболочечных, но не безоболочечных вирусов);
подверженность процесса инактивации (элиминации) вирусов небольшим вариациям параметров будет влиять на степень пригодности этапа;
пределы чувствительности методик количественного определения.
На основании совокупной оценки вышеперечисленных факторов принимается решение об отнесении этапа к эффективному, умеренно эффективному или неэффективному по способности к инактивации (элиминации) вирусов.
6.2. Частные варианты интерпретации данных по эффективности отдельного этапа процесса инактивации (элиминации) вирусов определяются следующими условиями:
если на этап процесса привнесено 6,0 lg вируса и из внесенного количества обнаружено 4,0 lg вируса, этап нельзя признать эффективным, хотя он и может вносить вклад в общую элиминацию;
если на этап процесса привнесено 6,0 lg вируса, однако вследствие цитотоксичности препарата предел чувствительности методики в препарате составляет 4,0 lg вируса, подтверждается лишь элиминация 2,0 lg вируса, а этап не признается эффективным. Этап процесса фактически способен элиминировать большее количество вируса, что можно подтвердить с помощью другого дизайна эксперимента;
если на этап процесса привнесено 6,0 lg вируса и из внесенного количества обнаружено 2,0 lg вируса, элиминировано значительное количество вируса. Препарат не является вирусологически стерильным. Однако если такое снижение содержания вирусов воспроизводимо и на него не влияют переменные процесса, процесс обладает определенной эффективностью. Он вносит вклад в совокупное снижение вирусной нагрузки и может признаваться в качестве процесса, снижающего вирусную нагрузку.
если на этап процесса привнесено 6,0 lg вируса и вирус не обнаруживается в препарате с пределом чувствительности, равным 2,0 lg вируса, подтверждена элиминация приблизительно 4,0 lg вируса. Этот показатель является значимым, а процесс фактически способен гарантированно элиминировать гораздо большее количество вируса, чем можно определить при количественном исследовании или заявить в спецификации;
если вирус подвергается инактивации, важна кинетика снижения инфекционности. Если этап процесса предполагает продолжительную инкубацию (например, нагревание в течение десяти часов), а инфекционность быстро достигает пределов обнаружения, процесс, вероятнее всего, обладает большим, чем зачастую можно подтвердить, вирулицидным эффектом. Однако если инфекционность снижается медленно, а пределы обнаружения достигаются к концу обработки, этап дает меньшую гарантию вирусной безопасности.
6.3. Процессы разделения в целом не являются эффективными этапами элиминации вирусов, однако они могут вносить вклад в их элиминацию. Процессы разделения, как правило, обладают рядом переменных, которые сложно поддаются контролю и разукрупнению для валидационных целей. Разделение зависит от крайне специфичных физико-химических свойств вируса, влияющих на его взаимодействие с гелевыми матрицами, и особенностей преципитации. Таким образом, разделение "модельного" вируса может полностью не соответствовать профилю разделения целевого вируса в связи с относительно небольшими различиями в таких поверхностных свойствах, как гликозилирование. Даже "релевантный" вирус, полученный в лаборатории, может проявлять в этом отношении другие свойства по сравнению с "диким" вирусом. Однако если процесс разделения дает воспроизводимое снижение вирусной нагрузки, и если параметры производства, влияющие на разделение, можно должным образом охарактеризовать и контролировать, а желаемую фракцию можно надежно отделить от предположительно содержащей вирусы фракции, тогда этот процесс может подпадать под критерии эффективного этапа.
6.4. Цель валидации заключается в выявлении эффективных этапов инактивации (элиминации) вирусов и получении оценки общей способности процесса производства инактивировать (элиминировать) их. Совокупный фактор снижения вирусной нагрузки, как правило, выражается суммой отдельных факторов в соответствии с приложением N 2 к настоящей главе. Простое суммирование малых факторов снижения, получаемых на каждом этапе, может вводить в заблуждение. Снижение вирусного титра на 1,0 lg и менее является ненадежным вследствие имеющихся ограничений валидационных исследований на очистку от вирусов, поэтому все факторы с такой величиной снижения вирусного титра следует игнорировать. Производители должны дифференцировать эффективные этапы от этапов процесса, которые могут вносить вклад в элиминацию, но обладают меньшей надежностью. Необходимо также учесть вероятность резистентности вируса, выжившего на одном этапе, к последующему этапу или, наоборот, наличия у него повышенной восприимчивости. В целом один этап, обладающий большим эффектом, дает большую гарантию вирусной безопасности, чем несколько этапов, обладающих таким же совокупным эффектом.
6.5. Если с помощью процесса производства достигается небольшое снижение вирусной нагрузки и основным фактором безопасности препарата является элиминация вируса, необходимо предусмотреть специфический дополнительный этап или этапы инактивации (элиминации).
6.6. В отношении всех вирусов производители должны обосновать приемлемость достигнутых факторов снижения вирусной нагрузки. Результаты будут рассматриваться в индивидуальном порядке.
6.7. Необходимо строго соблюдать принципы правил надлежащей производственной практики Союза, утверждаемых Комиссией, предусматривающие разграничение материала, подвергшегося эффективному этапу инактивации (элиминации) вирусов, и необработанного материала.
7. Ограничения валидационных исследований
Валидационные исследования вносят вклад в обеспечение установления приемлемого уровня безопасности лекарственного препарата, но самостоятельно не обеспечивают безопасность лекарственного препарата. Ряд факторов планирования и проведения валидационных экспериментов по очистке от вирусов может привести к некорректной оценке способности процесса элиминировать естественную вирусную инфекционность. К ним относятся перечисленные ниже факторы.
7.1. Различные лабораторные штаммы вируса могут отличаться по своей чувствительности к одному и тому же виду обработки. Таким образом, определенный выбранный в исследовании вирус может не отражать свойства того вируса, для моделирования процесса очистки от которого он выбран. Природные вирусы могут обладать непредсказуемыми свойствами, например, при взаимодействии с липидами, которое могут влиять на их свойства. Вирусные препараты, используемые в целях валидации процесса производства, чаще всего, получают на культуре тканей. Поведение вируса из тканевой культуры на производственном этапе может отличаться от поведения природного вируса, например, если природный и культивированный вирусы отличаются по чистоте или степени агрегации. Необходимо документировать штаммы вируса, их культивирование и количественное определение, а также пробоподготовку и хранение.
7.2. В некоторых случаях сложение логарифмических факторов снижения вирусной нагрузки недопустимо. Например, если матрица способна адсорбировать 104 инфекционных единиц вируса и далее неспособна адсорбировать материал с сопоставимой аффинностью, будут удалены все вирусы, введенные в количестве 104 инфекционных единиц, но только 1% вирусов при введении 106 инфекционных единиц. Таким образом, измеряемый клиренс будет розниться в зависимости от введенного титра.
7.3. Инактивация вирусной инфекционности зачастую представляет собой двухфазную кривую с быстрой начальной фазой и более медленной последующей фазой. Нельзя исключать, что вирус, избежавший первого этапа инактивации, будет более устойчив к последующим этапам. Как следствие совокупный фактор снижения вирусной нагрузки необязательно является суммой факторов снижения, рассчитанных на каждом этапе, на котором привносилась суспензия свежеприготовленного вируса. Например, если резистентная фракция принимает форму вирусных агрегатов, инфекционность может быть устойчива к различным видам химической обработки и нагреванию.
7.4. Обработка в модельном масштабе, как правило, будет отличаться от полномасштабной обработки, несмотря на проведение разукрупненного процесса.
7.5. Наличие антител к природному вирусу может влиять на их отделение от вирусной частицы или его чувствительность к химической инактивации, но оно также может осложнять планирование исследования за счет нейтрализации инфекционности. Определение правильности дизайна исследования может быть затруднительным. Содержание антител может являться значимой переменной процесса.
7.6. Небольшие различия в таких производственных параметрах, как содержание белка или температура, могут приводить к большим различиям в снижении вирусной инфекционности за счет различных механизмов.
8. Повторные исследования
8.1. Изменение процесса производства может потребовать проведения нового валидационного исследования.
8.2. По мере накопления теоретических знаний процессы валидации очистки будут требовать перепроверки в целях подтверждения того, что они продолжают удовлетворять приемлемому стандарту.
Приложение N 1
к главе 4 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
УКАЗАНИЯ
ПО СТАТИСТИЧЕСКОЙ ОЦЕНКЕ ВИРУСНЫХ ТИТРОВ И ФАКТОРОВ
СНИЖЕНИЯ ИХ СОДЕРЖАНИЯ, А ТАКЖЕ ОЦЕНКЕ ИХ ВАЛИДНОСТИ
1. Титрование вирусов подвержено вариации, что характерно для всех биологических систем количественного определения. Необходимо обеспечить правильность оценки вирусных титров и факторов снижения, полученных на их основе, а также валидность методик, чтобы определить надежность исследования. Цель статистической оценки заключается в установлении того, что исследование проведено на приемлемом уровне вирусологической компетентности.
2. Методы количественного определения могут быть квантовыми и количественными. К квантовым методам относятся методики определения инфекционности у животных и методики определения инфекционной дозы для тканевой культуры (TCID), в которых определяется число инфицированных и неинфицированных животных или клеточных культур. Титр инфекционности определяют как долю инфицированных животных или культур. В количественных методах измеренная инфекционность непрерывно варьирует в зависимости от количества введенного вируса. К количественным методам относятся методики бляшкообразования, в которых каждая бляшка соответствует одной инфекционной единице. Как квантовые, так и количественные методики подлежат статистической оценке.
3. Вариабельность методики может быть обусловлена ошибками разведения, статистическими эффектами и различиями в системе, которые неизвестны либо сложно поддаются контролю. Эти эффекты, будут более выражены при сравнении различных аналитических циклов (вариабельность между аналитическими циклами), чем при сравнении результатов одного аналитического цикла между собой (вариабельность внутри цикла).
4. 95% доверительные границы вариабельности внутри цикла и между циклами должны быть  0,5 lg или меньше. Вариабельность между аналитическими циклами следует контролировать путем включения собственного стандартного препарата, оценка активности которого должна находиться приблизительно в пределах
0,5 lg или меньше. Вариабельность между аналитическими циклами следует контролировать путем включения собственного стандартного препарата, оценка активности которого должна находиться приблизительно в пределах  0,5 lg от средней оценки, установленной в лаборатории в качестве приемлемой для применяемой методики. Вариабельность внутри цикла можно определять с помощью стандартных методов, описанных в руководствах по аналитическим исследованиям. При достаточном обосновании могут быть приняты результаты любого эксперимента, даже если прецизионность титрования меньше указанных целевых значений.
0,5 lg от средней оценки, установленной в лаборатории в качестве приемлемой для применяемой методики. Вариабельность внутри цикла можно определять с помощью стандартных методов, описанных в руководствах по аналитическим исследованиям. При достаточном обосновании могут быть приняты результаты любого эксперимента, даже если прецизионность титрования меньше указанных целевых значений.
5. Снижение вирусной нагрузки необходимо рассчитать на основании экспериментально установленных вирусных титров. По возможности необходимо определить 95% доверительные границы факторов снижения. Приблизительно их можно рассчитать с помощью следующей формулы:
 ,
,
где:
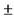 s - 95% для содержания вирусов в исходном материале;
s - 95% для содержания вирусов в исходном материале;
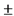 a - 95% границы для содержания вирусов в материале после прохождения этапа инактивации (элиминации).
a - 95% границы для содержания вирусов в материале после прохождения этапа инактивации (элиминации).
Если после этапа инактивации (элиминации) инфекционность в образце не обнаруживается, фактор снижения невозможно рассчитать статистическими методами. В целях получения оценки минимального фактора снижения титр необходимо принять равным одной инфекционной единице или менее в объеме с наибольшей испытанной концентрацией. Отсутствие признаков инфекционности в пробе будет характерно после применения мощных процессов инактивации. Чтобы максимально увеличить расчетный минимальный фактор снижения вирусной нагрузки с помощью эффективного процесса инактивации, необходимо отбирать как можно большее количество обработанного неразведенного материала.
Приложение N 2
к главе 4 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
УКАЗАНИЯ ПО РАСЧЕТУ ФАКТОРОВ СНИЖЕНИЯ ВИРУСНОЙ НАГРУЗКИ
Фактор снижения вирусной нагрузки (R) отдельного этапа инактивации или элиминации задается следующей формулой:
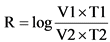 ,
,
где:
R - фактор снижения вирусной нагрузки;
V1 - объем исходного материала;
T1 - концентрация вируса в исходном материале;
V2 - объем материала после этапа инактивации (элиминации);
T2 - концентрация вируса после этапа инактивации (элиминации).
Данная формула учитывает как титр, так и объем материала до и после этапа инактивации (элиминации).
Факторы снижения, как правило, выражают в виде логарифмов, это значит, что даже при существенном снижении вирусной инфекционности она никогда не будет равна нулю. Согласно требованиям статьи (монографии) "Методы приготовления стерильных препаратов" фармакопеи Союза, утверждаемой Комиссией, в отношении методов стерилизации достаточными признаются процессы, которые обеспечивают гарантированный уровень стерильности (ГУС), равный 10-6 или менее, в отношении бактерий, плесеней и дрожжей. ГУС, равный 10-6, обозначает вероятность содержания не более одного жизнеспособного микроорганизма на 1 x 106 стерилизованных единиц лекарственного препарата.
Глава 5.1. производство и контроль качества
биотехнологических лекарственных препаратов, полученных
методом рекомбинантной ДНК
1. Область применения
Исследования в области молекулярной генетики и химии нуклеиновых кислот сделали возможными выявление, детальный анализ, передачу между организмами и экспрессию при заданных условиях (для синтеза полипептидов) генов, кодирующих естественные, биологически активные белки.
Многие лекарственные препараты, которые ранее было трудно получить из природных источников, сегодня можно получать с использованием технологии рекомбинантной ДНК. Кроме того, возможность синтезировать нуклеиновые кислоты и осуществлять над ними манипуляции позволяет конструировать гены, кодирующие модифицированные продукты, обладающие отличными от их природных аналогов свойствами, или даже принципиально новые продукты.
Общая стратегия при разработке препаратов с помощью технологии рекомбинантной ДНК - внедрение естественных, или намеренно модифицированных естественных последовательностей или новых нуклеотидных последовательностей в вектор, вводимый в подходящий организм-хозяин, для обеспечения эффективной экспрессии целевого продукта. В настоящее время разработаны и применяются прокариотические и эукариотические экспрессирующие системы "вектор/клетка-хозяин". На экспрессию чужеродных генов, вводимых в новый организм с помощью подходящего вектора, влияет комплекс факторов, поэтому важным аспектом разработки препарата является стабильная управляемая экспрессия клонированных ДНК-последовательностей.
В целях контроля качества этих препаратов необходимо придерживаться гибкого подхода, чтобы по мере накопления опыта производства и применения, а также вследствие разработки новых технологий, выполнять модификацию приведенных требований. Выполнение этих требований в отношении отдельных препаратов должно отражать их предполагаемое клиническое применение.
Цель данной главы - упростить сбор и представление данных, в составе регистрационного досье лекарственных препаратов на основе полипептидов, получаемых по технологии рекомбинантной ДНК и предназначенных для медицинского применения в Союзе. Требования настоящей главы связаны также с требованиями других актов в сфере обращения лекарственных средств, входящих в право Союза.
2. Вопросы производства
Производители лекарственных препаратов, получаемых по технологии рекомбинантной ДНК, должны соответствовать требованиям, предъявляемым правилами надлежащей производственной практики Союза, утверждаемых Комиссией, законодательству государств-членов о генетически модифицированных организмах, а также соответствовать общим требованиям по контролю качества биологических лекарственных препаратов.
Необходимо обеспечить должное качество всех реагентов, используемых при производстве лекарственных препаратов, включая компоненты среды культивирования, спецификации на них необходимо включить в регистрационное досье, они должны соответствовать всем действующим требованиям, установленным актами, входящими в право Союза, (например, требованиям по минимизации риска передачи возбудителя губчатой энцефалопатии через лекарственные препараты). Соответствующие спецификации следует включать в регистрационное досье на лекарственный препарат.
Испытания на активность, пирогены, стерильность и т.д., проводимые на препаратах, получаемых с помощью традиционных методов, также применимы к препаратам, получаемым по технологии рекомбинантной ДНК. При производстве препаратов не рекомендуется использовать агенты, способные вызывать сенсибилизацию у некоторых лиц (например, пенициллин или другие 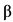 -лактамные антибиотики).
-лактамные антибиотики).
Несмотря на важность всесторонней характеристики лекарственного препарата, следует уделить большое внимание внутрипроизводственному контролю. Эта концепция доказала свою высокую эффективность при контроле качества бактериальных и вирусных вакцин, произведенных традиционными способами.
Некоторые факторы могут негативным образом отразиться на постоянстве качества, безопасности и эффективности препаратов, полученных по технологии рекомбинантной ДНК, поэтому таким факторам следует уделить особое внимание. При этом необходимо учитывать следующее:
все биологические системы претерпевают генетические изменения посредством мутаций и селекции, а чужеродные гены, вводимые в новые клетки хозяина, могут проявлять повышенную генетическую нестабильность. Цель молекулярно-генетических исследований показать, что правильная последовательность была получена и введена в клетку хозяина и что структура и число копий введенной последовательности остаются в клетке неизменными в ходе культивирования до завершения производства. Такие исследования могут дать значимую информацию, которую следует рассматривать в сочетании с результатами исследований целевого белка для обеспечения качества и постоянства свойств препарата;
целевые продукты, экспрессируемые чужеродными клетками-продуцентами, могут структурно, биологически или иммунологически отличаться от своих природных аналогов. Такие изменения могут возникнуть на посттрансляционном уровне или в ходе производства либо очистки и приводить к нежелательным клиническим последствиям. Должно быть показано, что наличие этих изменений допустимо, то есть не приводит к проявлению нежелательных клинических эффектов. В связи с этим присутствие целевых продуктов с такими изменениями необходимо обосновать и подтвердить наличие постоянного контроля;
выбор технологии производства влияет на свойства, разнообразие и количество потенциальных примесей в лекарственном препарате, а также определяет, в отношении каких процессов очистки необходимо подтверждать способность этих процессов элиминировать примеси (например, эндотоксины в препаратах, получаемых в бактериальных клетках, посторонние агенты и ДНК в препаратах, получаемых в клетках млекопитающих);
нежелательная изменчивость культуры в ходе производства может привести к изменениям, благоприятствующим экспрессии других генов в системе "хозяин-вектор", или вызывающим нарушения свойств препарата. Такая изменчивость может привести к изменению самого препарата (например, характера и степени гликозилирования) или его выхода и (или) может стать причиной возникновения количественных и качественных различий в профиле примесей. Именно поэтому обязательны процедуры, обеспечивающие постоянство условий производства, а также характеристик лекарственного препарата;
по мере перехода от лабораторной разработки к полномасштабному промышленному производству происходит существенное масштабирование процессов ферментации и (или) очистки, что может значительно повлиять на качество продукта, включая воздействие на его пространственную структуру, выход и (или) количественные и качественные различия в примесях. В связи с этим в ходе каждого производственного цикла необходимо предусмотреть внутрипроизводственный контроль и испытания по выпускающему контролю качества для подтверждения неизменности свойств получаемого препарата.
Настоящая глава применима ко всем препаратам, получаемым с использованием технологии рекомбинантной ДНК. Для отдельных препаратов могут возникнуть специфичные затруднения при контроле качества, поэтому при производстве и контроле качества каждого препарата следует учитывать все его свойства.
3. Генетическая разработка
Генетическая разработка должна соответствовать не только приведенным ниже правилам, но и требованиям, изложенным в главе 5.2 настоящих Правил.
3.1. Целевой ген, вектор и клетка-хозяин
Следует представить подробное описание клонированного гена. Это описание должно включать данные о его происхождении, подлинности и выделении, а также данные о происхождении и структуре экспрессирующего вектора. Необходимо представить описание штамма-хозяина или клеточной линии, включая историю исходного штамма или клеточной линии, их идентификационные признаки и потенциальные вирусные контаминанты. Особое внимание необходимо уделить возможности перекрестной контаминации другими клетками или вирусами.
3.2. Экспрессирующая конструкция
Следует представить подробные сведения о нуклеотидной последовательности целевого гена и фланкирующих контрольных участков экспрессирующего вектора, чтобы подтвердить, что конструкция гена идентична желаемой. Следует подробно описать этапы сборки экспрессирующей конструкции. Следует представить подробную карту и полную аннотированную последовательность функционально значимых участков вектора с указанием участков, секвенированных при создании конструкции, и участков, последовательность которых была определена на основе литературных данных. Последовательность в области точек лигирования фрагментов (непосредственно влияющих на экспрессию введенного гена) в ходе сборки конструкции необходимо подтвердить секвенированием. Необходимо идентифицировать все известные экспрессирующиеся последовательности.
3.3. Состояние рекомбинантной ДНК в клетке хозяина
Следует описать метод, с помощью которого вектор введен в клетку хозяина, и состояние рекомбинантной ДНК в ней (интегрированная или внехромосомная, число копий и т.д.). В отношении внехромосомных экспрессирующих систем необходимо определить процент клеток, сохраняющих экспрессирующую конструкцию. Последовательность экспрессирующей конструкции, кодирующую рекомбинантный препарат, необходимо проверить на уровне банка клеток. В системах, содержащих множественные интегрированные копии гена, полученные в результате амплификации или без нее, в дополнение к секвенированию молекул мРНК или кДНК следует провести детальное исследование с использованием разных рестрикционных ферментов и Саузерн-блоттинга, чтобы представить убедительные данные о целостности экспрессирующегося гена (генов).
3.4. Экспрессия
Следует детально описать стратегию обеспечения и контроля экспрессии соответствующего гена в ходе производства.
3.5. Стабильность экспрессирующей системы
Стабильность генетических и фенотипических характеристик системы "хозяин-вектор" необходимо исследовать до и после достижения уровня удвоения популяции или числа генераций, используемых при рутинном производстве (клетки предельного для производства клеточного возраста in vitro). Экспрессирующую конструкцию следует анализировать в клетках предельного для производства клеточного возраста in vitro, по меньшей мере, 1 раз для каждого главного банка клеток.
По результатам исследований стабильности необходимо также получить подробную информацию о:
числе копий гена относительно производительности культуры;
делециях и (или) вставках, влияющих на любую часть экспрессирующего вектора;
произведенном белке.
Анализ следует выполнять таким образом, чтобы его результаты могли подтвердить то, что число вариантов продукта ниже допустимого предела, устанавливаемого в индивидуальном порядке в зависимости от природы препарата и его предлагаемого применения. Можно провести анализ на уровне белка и (или) ДНК. Какой бы метод ни использовался, он должен пройти валидацию и для него должен быть определен предел обнаружения.
4. Контроль банков клеток
Необходимо следовать требованиям, изложенным в главе 1 настоящих Правил.
5. Ферментация или культивирование клеток
Необходимо представить четкое определение серии продукта, который подвергнется дальнейшей обработке.
Следует представить подробные данные по ферментации или культивированию клеток с описанием внутрипроизводственного контроля. Необходимо определить критерии отбраковки сбора клеток и преждевременного прекращения культивирования.
Наличие, степень и характер любой микробной контаминации в сосудах для культивирования следует внимательно изучить на соответствующей стадии в конце каждого производственного цикла. Необходимо представить подробную информацию для подтверждения достаточной чувствительности методов, используемых для выявления контаминации, и необходимо установить допустимые пределы контаминации.
В идеальном случае одновременно в одной производственной зоне следует культивировать не более одной клеточной линии. При параллельном культивировании других линий клеток следует документировать данные о разных клеточных линиях и представить результаты валидации, подтверждающие отсутствие (невозможность) перекрестной контаминации.
5.1. Производство с однократным сбором
Следует определить максимально допустимое число пассажей или удвоений популяции клеток при производстве. При определении этих показателей необходимо основываться на данных по стабильности системы "клетка-хозяин-вектор" на предельном для производства клеточном возрасте in vitro и превышающим его. Следует представить данные о постоянстве роста культуры и об обеспечении выхода препарата в определенных пределах. В конце производственных циклов следует проводить мониторинг характеристик клетки-хозяина и вектора. Необходимо представить подтверждение того, что вариабельность выхода препарата укладывается в установленные пределы и основные свойства и качество препарата остаются неизменными по ряду специфичных параметров.
5.2. Производство с многократным сбором
Следует определить период непрерывного культивирования, основываясь на данных о стабильности системы и постоянстве качества продукта в рамках этого периода и в течение большего периода времени. На протяжении культивирования требуется мониторинг системы производства. Требуемая частота и тип мониторинга зависят от нескольких факторов, включая тип экспрессирующей системы и препарата, а также общую продолжительность периода непрерывного культивирования. Приемлемость сборов для последующей обработки должна строго зависеть от использующегося режима мониторинга. Необходимо представить подтверждение того, что выход препарата укладывается в установленные пределы и основные свойства и качество препарата остаются неизменными по ряду специфичных параметров.
6. Очистка продукта
6.1. Методы
Следует подробно описать, обосновать и валидировать методы, используемые для очистки продукта и применяемого внутрипроизводственного контроля, включая заданные в спецификациях допустимые пределы. Использование методик, включающих аффинную хроматографию (например, с использованием моноклональных антител) должно сопровождаться надлежащими мерами, обеспечивающими отсутствие негативного влияния этих веществ и любых других потенциальных контаминантов, образующихся при их использовании, на качество и безопасность лекарственного препарата. Следует учитывать требования, содержащиеся в главе 10 настоящих Правил и главе 4 настоящих Правил по валидации методов обеспечения вирусной безопасности.
Следует тщательно определить критерии повторной переработки всех промежуточных продуктов и готового нерасфасованного продукта, провести валидацию и обосновать их применимость.
6.2. Валидация процедуры очистки
Следует тщательно проанализировать возможности процесса очистки элиминировать (инактивировать) нежелательные белки, нуклеиновые кислоты, углеводы, вирусы, происходящие из клеток хозяина, и другие примеси, включая родственные белки.
Следует провести исследование с использованием тщательно отобранной группы вирусов, обладающих широким диапазоном физико-химических свойств, значимых для их поведения в ходе очистки в соответствии требованиями главы 4 настоящих Правил и намеренно привнесенных в неочищенный продукт (spiking). Следует также подтвердить способность процесса очистки элиминировать (инактивировать) специфичные контаминанты, такие как белки клетки-хозяина, ДНК и другие потенциальные производственные примеси. При необходимости с этой целью можно использовать такие контаминанты в концентрациях, превышающих ожидаемую в ходе нормального производства (spiking). Следует определить фактор снижения содержания таких контаминантов на каждой стадии очистки и общий фактор снижения их содержания.
Валидация процесса очистки должна также включать обоснование рабочих условий, например, емкости колонки, регенерации и санитарной обработки колонок и продолжительности использования колонок. Колонки также следует подвергать валидации в отношении вымывания лигандов (например, красителя, аффинного лиганда и др.) и (или) хроматографического субстрата в течение ожидаемого срока службы колонки.
7. Активная фармацевтическая субстанция
7.1. Установление характеристик активной
фармацевтической субстанции
7.1.1. Физико-химическая характеристика, относительная молекулярная масса, изоэлектрическая точка.
Необходимо тщательно установить характеристики активной фармацевтической субстанции с помощью физико-химических и биологических методов. Особое внимание следует уделить использованию широкого диапазона аналитических методик, оценивающих разные физико-химические свойства молекулы; например, размер, заряд, изоэлектрическую точку и гидрофобность. Перечень аналитических возможностей не входит в настоящую главу. Далее представлены отдельные требования к разновидностям анализа.
7.1.2. Подтверждение структуры активной фармацевтической субстанции (включая сравнение с стандартным материалом или природным продуктом).
Следует представить данные об аминокислотной последовательности продукта гена в объеме, достаточном для его адекватной характеристики. Необходимая степень подробности описания генетической последовательности зависит от размера и сложности молекулы, а также от использования других испытаний для установления характеристик. Во многих случаях для оценки последовательности возможно использование разделения с помощью ВЭЖХ с последующим секвенированием пептидов, полученных с помощью ферментативного расщепления. Следует обратить внимание на возможное наличие N-концевого метионина и N-формилметионина, сигнальных или лидерных последовательностей, других возможных N и C-концевых модификаций (протеолитический процессинг). Следует рассмотреть возможность использования современных методов масс-спектрометрии при характеристике первичной структуры.
7.1.3. Посттрансляционные модификации
Помимо протеолитического процессинга потенциальными типами посттрансляционных модификаций являются N и O-гликозилирование и, например, ацетилирование, гидроксилирование и 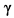 -карбоксилирование. Кроме того, существуют посттрансляционные модификации, возникающие в результате процессов расщепления, например, дезаминирования и окисления.
-карбоксилирование. Кроме того, существуют посттрансляционные модификации, возникающие в результате процессов расщепления, например, дезаминирования и окисления.
Некоторые препараты рекомбинантных ДНК являются гликопротеинами. Существует огромное множество олигосахаридных структур, которые характеризуются гетерогенностью гликоформ как в их естественной форме, так и образующихся по технологии рекомбинантных ДНК. На характер этой гетерогенности могут влиять многие факторы. Профиль гликозилирования молекулы может играть важную роль при определении ее активности особенно in vivo. Объем проводимого анализа должен определяться ролью, которую играют углеводные группы, если эти данные известны. Следует рассмотреть весь диапазон разных аналитических методик (например, количественное изоэлектрическое фокусирование или капиллярный электрофорез, анионообменная хроматография для анализа моносахаридного компонента и определения олигосахаридов, лектин-аффинная хроматография и масс-спектрометрия).
7.1.4. Данные о конформации макромолекул.
Рекомендуется применять соответствующие испытания, позволяющие установить то, что продукт имеет желаемую конформационную структуру и степень агрегированности. Примерами подходящих для таких целей методов являются электрофорез в полиакриламидном геле, изоэлектрическое фокусирование, высокоэффективная жидкостная хроматография (эксклюзионная, обращенно-фазовая, ионообменная, аффинная), пептидное картирование с последующим секвенированием, светорассеяние, ультрафиолетовая спектроскопия, круговой дихроизм и масс-спектроскопия. Значимая информация получается при проведении дополнительных исследований продукта, используя, например, ядерно-магнитную резонансную спектроскопию, рентгеновскую кристаллографию и соответствующие иммунохимические методы.
7.1.5. Установление биологических и иммунологических характеристик молекул, способ выражения дозировки продукта.
Для биологической и иммунологической характеристики следует использовать как можно более широкий набор методов. Необходимо определить удельную активность высокоочищенного материала (единицы активности на массу продукта).
По возможности биологическую активность продукта и его физические характеристики, включая аминокислотную последовательность, следует сравнивать с аналогичными показателями высокоочищенного продукта природного происхождения.
7.2. Чистота
Необходимо представить данные о контаминантах, наличие которых ожидается в готовом обработанном продукте. Необходимо обосновать допустимую степень контаминации и представить критерии приемлемости или отбраковки промышленной серии. Важно оценивать чистоту препарата с использованием как можно более широкого диапазона методик, включая физико-химические и иммунологические. Необходимо провести испытания на наличие нежелательных материалов, проистекающих от клеток хозяина, а также материалов, которые могли быть использованы (добавлены) в процессе производства или очистки, и, если применимо, контаминации вирусами и нуклеиновыми кислотами.
8. Постоянство характеристик и посерийный контроль готовой
нерасфасованной (балк) фармацевтической субстанции
В целях установления постоянства характеристик подлинности, чистоты и активности необходимо провести всесторонний анализ начальных серий продукта. Впоследствии можно будет ограничиться сокращенным набором испытаний, который представлен ниже. Следует четко различать аналитические испытания, проводимые в ходе разработки препарата с целью полного установления характеристик фармацевтической субстанции, и испытания, рутинно выполняемые с каждой серией очищенного нерасфасованного продукта.
8.1. Постоянство характеристик
Число последовательных серий нерасфасованного обработанного продукта, подвергшихся анализу, должно составлять не менее 5 серий (при достаточном обосновании допустимо меньшее число серий). Их следует охарактеризовать настолько полно, насколько это возможно, чтобы подтвердить постоянство состава. При производстве с многократным сбором продукта, как правило, следует исследовать серии продукта из разных циклов культивирования. Следует использовать биологические, физико-химические и иммунологические методы для характеристики и анализа активной фармацевтической субстанции (включая методы, подтверждающие постоянство профиля гликозилирования гликопротеинов) и методы выявления и идентификации примесей. Все выявленные различия между сериями необходимо документировать.
8.2. Посерийный контроль качества
8.2.1. Подлинность.
Для подтверждения подлинности каждой серии препарата необходимо выбрать часть испытаний, использованных для характеристики очищенной активной фармацевтической субстанции в соответствии с указания раздела 7.1 настоящей главы. Используемые методы должны включать испытания физико-химических и иммунологических свойств вместе с испытанием на ожидаемую биологическую активность. В зависимости от масштабов других испытаний на подлинность следует проводить верификацию N- или C-концевой аминокислотной последовательности или использовать другие методы, например, пептидное картирование.
8.2.2. Чистота.
Степень желаемой и достижимой чистоты будет зависеть от нескольких факторов: природы и целевого назначения продукта, метода его производства и очистки, а также от степени постоянства характеристик производственного процесса. Используя современные технологические процессы, можно добиться очень высокой степени чистоты большинства препаратов.
Следует определять чистоту продукта в каждой серии, она должна оставаться в заданных пределах. Анализ должен включать чувствительные и надежные методы исследования ДНК клетки-хозяина и (или) вектора, которым необходимо подвергать каждую серию продукта, приготовленного из линий клеток млекопитающих, при этом необходимо установить верхнее предельное содержание ДНК. Рекомендуется также проводить испытания каждой серии нерасфасованного продукта, полученного из других эукариотических клеточных систем, на содержание ДНК и установить предельное содержание ДНК. В прокариотических экспрессирующих системах следует проводить испытания на ДНК, если это необходимо для обеспечения качества препарата. Также необходимо с помощью достаточно чувствительного метода количественного определения определять остаточные клеточные белки путем проведения анализа с соответствующей чувствительностью (например, parts per million (ppm), частей на миллион) и установить строгое верхнее предельное их содержание. В некоторых случаях потенциальные примеси, например ДНК, могут быть выявлены в промежуточных продуктах очистки на более раннем этапе.
8.2.3. Испытание на активность.
Следует определить активность каждой серии препарата (например, в единицах биологической активности на миллилитр), по возможности используя соответствующий национальный или международный стандартный препарат, калиброванный в единицах биологической активности как это указано в разделе 9 настоящей главы.
Следует оценить удельную активность препарата (единицы биологической активности на единицу массы препарата). Оцененную удельную активность необходимо документировать. Для стандартизации определения удельной активности необходимо использовать высокоочищенный стандартный препарат в соответствии с разделом 9 настоящей главы.
Рекомендуется оценить корреляцию между результатами определения активности биологическими методами и физико-химическими методами анализа и представить полученные данные. По возможности серии следует стандартизовать, используя точные физико-химические методики, а биологический анализ следует применять для подтверждения биологической активности препарата "в указанных пределах".
9. Спецификация и стандартные материалы
Исследования, указанные в разделе 7 настоящей главы, позволяют подготовить окончательную спецификацию на препарат, если они подтверждаются данными, полученными при исследованиях последовательных серий, и данными посерийного контроля в соответствии с разделом 8 настоящей главы.
Правильно подобранную серию препарата, предпочтительно изученную клинически, следует полностью охарактеризовать с точки зрения ее химического состава, чистоты, активности и биологической активности, включая по возможности полное аминокислотное секвенирование. Исследованную таким образом серию препарата рекомендуется сохранить для использования в качестве химического и биологического стандартного материала.
Следует установить критерии продолжительности использования (срока годности) и (или) возможного повторного испытания и квалификации стандартных образцов.
10. Лекарственный препарат и фармацевтическая разработка
Разработку лекарственного препарата необходимо подробно описать и обосновать, уделив особое внимание описанию наличия и содержания стабилизатора (например, альбумина и (или) детергентов). Необходимо подтвердить, что лекарственный препарат в окончательных контейнерах соответствует требованиям фармакопеи Союза, утверждаемой Комиссией, и актов в сфере обращения лекарственных средств, входящих в право Союза. При невозможности выполнения испытаний производитель должен обосновать их исключение.
Определения
Для целей настоящей главы используются понятия (термины), которые означают следующее:
"готовая нерасфасованная фармацевтическая субстанция" - получаемый из нерасфасованного сбора по завершении процесса производства готовый продукт, который хранится в одном контейнере или при необходимости в нескольких одинаковых контейнерах и используется при производстве готовой лекарственной формы. Производство серии готового продукта должно быть четко описано и подробно документировано производителем;
"лекарственный препарат" - активная фармацевтическая субстанция в составе лекарственной формы, расфасованной в первичную упаковку (укупоренные контейнеры) для ее сохранности, которая является лекарственным препаратом. Контейнеры, составляющие одну серию фасовки, должны обрабатываться в рамках одного производственного цикла, их содержимое и биологическая активность должны быть однородны;
"нерасфасованный сбор" - однородный пул отдельных сборов или лизатов, обрабатываемый в ходе одного цикла очистки.
Глава 5.2. Анализ экспрессирующей конструкции клеток,
используемых в производстве белковых препаратов, полученных
по технологии рекомбинантной ДНК
1. Область применения
Настоящая глава содержит указания по определению характеристик экспрессирующей конструкции, предназначенной для синтеза белковых препаратов по технологии рекомбинантной ДНК в эукариотических и прокариотических клетках и данные, важные для оценки структуры экспрессирующих конструкций, используемых в производстве белков на основе рекомбинантной ДНК. Рассмотрение всех аспектов качества лекарственных препаратов, полученных по технологии рекомбинантной ДНК, не является целью настоящей главы.
Экспрессирующая конструкция - экспрессирующий вектор, содержащий последовательность, кодирующую рекомбинантный белок. Для обеспечения качества и постоянства свойств лекарственного препарата следует анализировать области экспрессирующих конструкций с использованием методов изучения нуклеиновых кислот в сочетании с подходами, применяемыми для анализа очищенных рекомбинантных белков. Анализ экспрессирующих конструкций на уровне нуклеиновых кислот является элементом целостного процесса оценки качества, при этом в таком исследовании оценивается только кодирующая последовательность рекомбинантного гена, но не точность трансляции или другие характеристики рекомбинантного белка, например, вторичная, третичная структуры и посттрансляционные модификации.
2. Обоснование анализа экспрессирующей конструкции
Цель анализа экспрессирующей конструкции - подтвердить, что в клетку-хозяина внедрена правильная кодирующая последовательность продукта и она поддерживается в ней в течение культивирования до окончания производственного процесса. Генетическая последовательность рекомбинантных белков, синтезируемая в живых клетках, может подвергаться мутациям, способным изменить свойства белка, что может привести к негативным последствиям у пациентов. Ни один экспериментальный подход не позволяет выявить одновременно все возможные модификации белков. Для изучения аминокислотной последовательности и структурных особенностей экспрессирующегося белка, обусловленного посттрансляционными модификациями, например, протеолитическим процессингом, гликозилированием, фосфорилированием и ацетилированием, допускается использование методов белкового анализа. Белковый анализ может не выявить все изменения структуры рекомбинантного белка, обусловленные мутациями в кодирующей его последовательности. В этом случае данные анализа нуклеиновых кислот могут представлять особое значение. Относительная важность анализа нуклеиновых кислот и белков для разных препаратов различается.
Анализ нуклеиновых кислот можно применять для верификации кодирующей последовательности, а также для оценки физического состояния экспрессирующей конструкции. Анализ нуклеиновых кислот проводят, чтобы убедиться, что экспрессируемый белок имеет правильную аминокислотную последовательность, однако этот метод не предназначен для обнаружения малых количеств вариантных последовательностей. Если клетки-продуценты содержат множество встроенных копий экспрессирующей конструкции, не все из которых транскрибируются, наиболее подходящим методом может быть исследование самих продуктов транскрипции при помощи анализа мРНК или кДНК, а не исследование геномной ДНК. Аналитические подходы, направленные на изучение всей совокупности нуклеиновых кислот, например, проводимые на пуле клонов или материале, амплифицированном при помощи полимеразной цепной реакции, можно рассматривать как альтернативу методам, зависящим от отбора отдельных клонов ДНК. Возможно использование других методик, позволяющих быстро и с достаточной чувствительностью верифицировать в экспрессирующей конструкции последовательность, кодирующую рекомбинантный белок.
В настоящей главе описываются данные, которые следует учитывать при составлении характеристики экспрессирующей конструкции в процессе разработки и валидации производственного процесса. Аналитические методики должны пройти валидацию для подтверждения способности обнаружения последовательности. Валидационная документация должна по меньшей мере включать расчет пределов обнаружения вариантных последовательностей, для этого необходимо провести валидацию методик секвенирования нуклеиновых кислот либо белков. Принципы и рекомендации по анализу, описанные в настоящей главе, необходимо регулярно пересматривать для учета новых технических достижений и научных данных.
3. Установление характеристик экспрессирующей системы
3.1. Экспрессирующая конструкция и клон клеток,
использованные для создания главного банка клеток
Производитель должен описать происхождение нуклеотидной последовательности, кодирующей белок. Это описание должно включать идентификацию и источник клеток, из которых получена последовательность нуклеотидов была исходно. Необходимо приложить описание методов, применявшихся для получения ДНК, кодирующей белок.
Необходимо представить подробное описание этапов сборки экспрессирующей конструкции. Это описание должно включать источник и функцию компонентов конструкции, например, ориджинов (точек начала) репликации, генов антибиотикорезистентности, промоторов, энхансеров, а также является ли белок продуктом слияния (химерным). Необходимо представить подробную карту компонентов и полную последовательность плазмиды с указанием участков, секвенированных в ходе создания конструкции, и участков, последовательность которых была взята из литературных данных. Необходимо указать другие экспрессирующиеся белки, кодируемые этой плазмидой. При помощи секвенирования ДНК необходимо определить нуклеотидную последовательность участка, кодирующего целевой ген, а также связанные с ним фланкирующие области, введенные в вектор, включая области в точках лигирования.
Следует представить описание метода введения экспрессирующей конструкции в клетку-хозяина. Кроме того, необходимо подробно описать методы, использованные для амплификации экспрессирующей конструкции, и критерии отбора клона клеток в качестве будущего продуцента.
3.2. Система банков клеток
Для синтеза рекомбинантных белков необходимо использовать только надлежащим образом охарактеризованные ГБК и РБК. Банк клеток представляет собой комплект хранящихся в определенных условиях контейнеров одинакового состава, каждый из которых содержит аликвоту, отобранную из одного и того же пула клеток. ГБК обычно получают из отобранного клона клеток, содержащего экспрессирующую конструкцию. РБК получают, культивируя клетки из одного или нескольких контейнеров ГБК. В регистрационном досье необходимо подробно описать процесс получения клеточной линии и формирования банка клеток, включая методы и реагенты, применявшиеся при культивировании, клеточный возраст in vitro и условия хранения. Во всех банках клеток должно быть определено наличие значимых генотипических и фенотипических маркеров, которые могут включать экспрессию рекомбинантного белка и наличие экспрессирующей конструкции.
Экспрессирующую конструкцию в ГБК следует анализировать согласно приведенной ниже последовательности испытаний. Если такой анализ на ГБК провести невозможно, его необходимо выполнить на каждом из рабочих банков.
Для определения числа копий экспрессирующей конструкции, выявления в ней инсерций или делеций, а также для установления числа участков интеграции следует применять картирование с использованием рестрикционных эндонуклеаз или другие подходящие методы. Для внехромосомных экспрессирующих систем следует определить процент клеток хозяина, сохраняющих экспрессирующую конструкцию.
Необходимо подтвердить (верифицировать) правильность последовательности экспрессирующей конструкции, кодирующей рекомбинантный белок. Для внехромосомных экспрессирующих систем следует изолировать экспрессирующую конструкцию и верифицировать нуклеотидную последовательность, кодирующую белок, не проводя дальнейшее клонирование. Для клеток с хромосомными копиями экспрессирующей конструкции нуклеотидную последовательность, кодирующую белок, следует подтверждать с помощью повторного клонирования и секвенирования хромосомных копий. В качестве альтернативного варианта нуклеотидную последовательность, кодирующую белок, можно верифицировать секвенированием пула клонов кДНК или материала, амплифицированного с помощью полимеразной цепной реакции. Кодирующая последовательность должна быть идентична (в границах предела обнаружения используемой методики) последовательности, требуемой для экспрессирующей конструкции, описанной в подразделе 3.1 настоящей главы, кроме того, она должна соответствовать ожидаемой аминокислотной последовательности белка.
3.3. Предельный для производства клеточный возраст in vitro
Предельный для производства клеточный возраст in vitro необходимо устанавливать на основе данных, полученных при культивировании клеток-продуцентов до предлагаемого клеточного возраста in vitro или в течение большего периода времени в рамках опытно-промышленного или промышленного производства. Как правило клетки-продуценты получают, культивируя клетки из РБК, тогда как ГБК для подготовки клеток-продуцентов следует применять только при надлежащем обосновании.
Экспрессирующую конструкцию клеток-продуцентов в ГБК необходимо проанализировать однократно (в соответствии с требованиями подраздела 3.2 настоящей главы). Последовательность экспрессирующей конструкции, кодирующей белок, в клетках-продуцентах может быть проверена при помощи анализа нуклеиновых кислот или анализа готового белкового препарата. Ранее установленный предельный для производства клеточный возраст in vitro можно увеличивать, только основываясь на данных, полученных на клетках, культивированных до клеточного возраста in vitro, равного или превышающего новый предельный клеточный возраст in vitro.
4. Заключение
Определение характеристик экспрессирующей конструкции и готового очищенного белка - важные этапы обеспечения постоянства производства препарата, изготавливаемого по технологии рекомбинантной ДНК. Оценка аналитических данных, полученных по результатам анализа нуклеиновых кислот и готового очищенного белка, необходима для обеспечения надлежащего качества рекомбинантных белковых препаратов.
5. Определения
Для целей настоящей главы используются понятия (термины), которые означают следующее:
"значимые генотипические и фенотипические маркеры" - маркеры, позволяющие идентифицировать штамм клеточной линии. Они должны включать экспрессию рекомбинантного белка или наличие экспрессионной конструкции;
"клеточный возраст in vitro" - период с момента оттаивания контейнеров с ГБК до сбора препарата в производственной емкости, измеряемый по продолжительности времени культивирования, степени удвоения популяции клеток или по числу пассажей клеток при субкультивировании с использованием определенной процедуры разведения культуры;
"опытно-промышленное производство" - получение рекомбинантного белка при помощи технологических процессов, полностью повторяющих или имитирующих полномасштабное промышленное производство. Методы культивирования клеток, сбора продукта и его очистки должны быть идентичными, за исключением масштаба производства;
"сайт интеграции" - участок, в котором в геном клетки встраивается одна или несколько копий экспрессирующей конструкции;
"фланкирующие контрольные области" - некодирующие нуклеотидные последовательности, прилегающие к 5'- и 3'-концам последовательности нуклеотидов нуклеиновой кислоты, кодирующей продукт. Фланкирующие контрольные области содержат важные элементы, влияющие на транскрипцию, трансляцию или стабильность кодирующей последовательности. Эти участки включают, например, промотор, энхансер и сплайсинговые последовательности и не содержат ориджины репликации и генов антибиотикорезистентности;
"экспрессирующая конструкция" - экспрессирующий вектор, содержащий последовательность, кодирующую рекомбинантный белок, и элементы, необходимые для ее экспрессии.
Глава 5.3. Доклиническая оценка безопасности лекарственных
препаратов, полученных с использованием биотехнологических
методов (основные требования)
1. Введение
1.1. Цели
Государства-члены используют гибкий, основанный на индивидуальном подходе, научно обоснованный принцип доклинической оценки безопасности лекарственных препаратов, полученных с использованием биотехнологических методов, необходимый для подтверждения правильности клинической разработки и возможности регистрации этих лекарственных препаратов.
Основные задачи доклинической оценки безопасности:
определение первоначальной безопасной дозы и схемы последующего увеличения дозы лекарственного препарата при введении человеку;
выявление потенциальных органов-мишеней, в отношении которых проявляется токсическое воздействие препарата, и установление обратимости проявлений токсичности;
определение параметров безопасности лекарственного препарата для их последующего мониторинга в ходе клинического исследования.
Соблюдение требований, изложенных в настоящей главе, необходимо для улучшения качества, согласованности проведения и оценки результатов доклинических исследований безопасности, сопровождающих разработку биологических лекарственных препаратов.
1.2. Область применения
Положения настоящей главы представляют описание базовой модели доклинической оценки безопасности лекарственных препаратов, полученных с использованием биотехнологических методов. Они применимы в отношении препаратов, полученных из охарактеризованных клеток, с помощью различных экспрессирующих систем, к которым относятся бактериальные и дрожжевые клетки, клетки насекомых, растений и млекопитающих. Предполагаемые показания к применению могут включать in vivo диагностику, лечение и профилактику. Активные фармацевтические субстанции представляют собой белки, пептиды, их производные и препараты, в состав которых они входят. Активные фармацевтические субстанции могут быть выделены из клеточных культур или получены с использованием технологии рекомбинантной ДНК, включая производство с помощью трансгенных растений и животных. К ним относят в том числе: цитокины, активаторы плазминогена, рекомбинантные факторы плазмы крови, факторы роста, гибридные белки (фьюжн белки, химерные белки), ферменты, рецепторы, гормоны и моноклональные антитела.
Требования настоящей главы, также применимы к вакцинам, полученным с использованием технологии рекомбинантной ДНК, химически синтезированным пептидам, препаратам, полученным из плазмы, эндогенным белкам, выделенным из тканей человека, лекарственным препаратам на основе олигонуклеотидов.
Положения настоящей главы не распространяются на антибиотики, экстракты аллергенов, гепарин, витамины, клеточные компоненты крови, традиционные бактериальные или вирусные вакцины, ДНК-вакцины, а также на препараты для клеточной и генной терапии, если в главах, регламентирующих изучение этих групп лекарственных препаратов не указано иное.
2. Спецификация на исследуемый материал
Риск, связанный с безопасностью применения биотехнологических лекарственных препаратов, может быть обусловлен присутствием в них примесей или контаминантов. Предпочтительнее удалять примеси и контаминанты с помощью процессов очистки, нежели полагаться на доклинические исследования для оценки их безопасности. В любом случае в целях надлежащего планирования доклинических исследований безопасности необходимо всегда всесторонне охарактеризовать свойства лекарственного препарата.
Существуют потенциальные риски, обусловленные наличием контаминантов из клетки-хозяина, которыми могут быть бактерии, дрожжи, клетки насекомых, растений или млекопитающих. Наличие контаминантов из клетки-хозяина может быть причиной развития аллергических или других иммунологических реакций. Теоретически существует опасность возникновения нежелательных реакций, обусловленных присутствием контаминантов в виде нуклеиновых кислот клетки-продуцента, в связи с их возможной интеграцией в геном хозяина. Для лекарственных препаратов, полученных с использованием клеток насекомых, растений и млекопитающих или клеточного материала из трансгенных растений и животных, необходимо учитывать дополнительный риск вирусной контаминации.
Как правило, препарат, изученный в основных фармакологических и токсикологических доклинических исследованиях, должен быть сопоставим с препаратом, предлагаемым для начальных клинических исследований. Однако необходимо учитывать, что во время осуществления программ исследований могут происходить изменения производственного процесса, направленные на совершенствование качества препарата и процесса его производства. При экстраполяции на человека данных, полученных в экспериментальных исследованиях на животных, необходимо рассмотреть возможное влияние изменений, внесенных в технологию производства, на свойства препарата и оценить потенциальную значимость подобных изменений, включая значимость посттрансляционных модификаций белка действующего вещества препарата.
Если в ходе программы разработки вводится новый производственный процесс, модифицируется существующий производственный процесс или происходят другие значимые изменения лекарственного препарата либо его состава, необходимо подтвердить сопоставимость характеристик изучаемого лекарственного препарата, полученного до и после изменения его технологического процесса. Сопоставимость может быть установлена по результатам сравнительной оценки биохимических и биологических свойств (то есть определения подлинности, чистоты, стабильности и активности). В отдельных случаях могут потребоваться дополнительные исследования (то есть исследования фармакокинетических параметров, фармакодинамических свойств и (или) безопасности). Необходимо представить научное обоснование используемого подхода для оценки сопоставимости препаратов. Информация об оценке сопоставимости препаратов при внесении изменений в процесс производства приведена в главах 9.1 - 9.2 настоящих Правил.
3. Доклинические исследования безопасности
3.1. Общие принципы
Целью доклинических исследований безопасности является определение фармакологического и токсического действия препарата, оцениваемого на протяжении всего периода клинической разработки, а не только на этапе предшествующем началу клинических исследований с участием человека. Для такой характеристики могут потребоваться исследования как in vivo, так и in vitro. При этом для биологических лекарственных препаратов, которые структурно и фармакологически сопоставимы с препаратом, уже широко применяемым в клинической практике, исследования токсичности могут быть менее объемными.
При проведении доклинической оценки безопасности лекарственного препарата необходимо в первую очередь учитывать следующее:
выбор соответствующих (релевантных) видов животных, наиболее чувствительных в отношении фармакологического и токсического действия;
возраст экспериментальных животных;
физиологическое состояние животных;
способ введения лекарственного препарата, включая расчет дозы, путь введения и режим дозирования;
стабильность исследуемого материала в условиях исследования.
Токсикологические исследования должны проводиться в соответствии с требованиями правил надлежащей лабораторной практики Союза, утверждаемых Комиссией. Отдельные исследования, требующие использования специфических тест-систем, которые часто необходимы при исследовании биотехнологических лекарственных препаратов, могут частично не соответствовать требованиям правил надлежащей лабораторной практики Союза, утверждаемых Комиссией. В этих случаях необходимо указать области несоответствия и оценить степень их значимости по отношению к общей оценке безопасности. В отдельных случаях отсутствие полного соответствия исследований токсичности требованиям правил надлежащей лабораторной практики Союза, утверждаемых Комиссией, не является строгим препятствием для перехода к проведению клинических исследований и последующей регистрации биотехнологического лекарственного препарата.
Традиционные подходы, используемые для исследования токсичности лекарственных препаратов, могут не в полной мере подходить для биологических лекарственных препаратов. Причиной этого являются уникальность и разнообразие структурных и биологических свойств биотехнологических лекарственных препаратов, в частности видоспецифичность, иммуногенность и непредсказуемые плейотропные эффекты.
3.2. Биологическая активность (фармакодинамика)
Допускается оценивать биологическую активность с использованием испытаний in vitro в целях определения клинически значимых эффектов лекарственного препарата. Возможно использование клеточных линий и (или) первичных клеточных культур для оценки непосредственного влияния препарата на фенотип клеток и их пролиферацию. Вследствие видоспецифичности многих биотехнологических лекарственных препаратов в целях изучения их токсичности необходимо выбрать релевантный вид животных. Для прогнозирования активности в экспериментах in vivo, а также количественной оценки относительной чувствительности различных видов животных, в том числе человека, к данному биологическому лекарственному препарату могут быть использованы эксперименты in vitro на клеточных линиях, полученных из клеток млекопитающих. Целью подобных исследований может являться определение, например, специфичности и аффинности связывания с рецептором и (или) фармакологических эффектов. Кроме того, на основании результатов этих исследований могут быть выбраны релевантные виды животных, которые будут использованы в дальнейших фармакологических и токсикологических исследованиях in vivo. Объединенные результаты исследований in vitro и in vivo позволяют провести экстраполяцию полученных экспериментальных данных на человека. Для обоснования предлагаемого применения лекарственного препарата в клинических исследованиях часто используют результаты исследований in vivo, оценивающие фармакологическую активность, включая определение механизма действия препарата.
При исследовании лекарственных препаратов моноклональных антител необходимо подробно описать их отличные от предполагаемых иммунологические свойства, в частности антигенную специфичность, способность связывать комплемент, а также непредусмотренную реактивность и (или) цитотоксичность по отношению к тканям человека. Такие исследования перекрестной реактивности должны быть проведены с использованием соответствующих иммуногистохимических методик и различных тканей человека.
3.3. Выбор видов животных (животной модели)
Наличие биологической активности, а также видовая и (или) тканевая специфичность многих лекарственных препаратов, полученных с использованием биотехнологических методов, часто препятствуют проведению стандартных исследований токсичности на широко используемых видах животных (например, крысах и собаках). Программы исследований безопасности должны включать использование релевантных видов животных. Релевантным является такой вид животных, при проведении исследования на котором изучаемый материал является фармакологически активным за счет взаимодействия препарата с рецептором (или эпитопом, если речь идет о моноклональных антителах). Для поиска релевантных видов животных могут быть использованы различные методы (например, иммунохимические или функциональные тесты). Сведения о распределении рецепторов и эпитопов позволяют получить более глубокое представление о потенциальной токсичности препарата в условиях in vivo.
При исследованиях моноклональных антител релевантными видами животных являются те, у которых экспрессируется необходимый эпитоп и для тканей которых можно продемонстрировать аналогичный профиль перекрестной реактивности по сравнению с тканями человека. Это позволяет правильно оценивать токсичность, обусловленную взаимодействием препарата с эпитопом либо неспецифической перекрестной тканевой реактивностью. Использование видов животных, у которых необходимый эпитоп не экспрессируется, может быть полезно для оценки токсичности в случае, если показана сравнимая неспецифическая перекрестная реактивность препарата с тканями человека.
Программа оценки безопасности, как правило, должна включать исследования на 2 релевантных видах животных. В отдельных обоснованных случаях достаточно использования 1 релевантного вида животных (например, если был найден только 1 релевантный вид или биологическая активность биотехнологического лекарственного препарата хорошо изучена). Кроме того, даже в случаях, когда для характеристики токсичности в краткосрочных исследованиях необходимо использовать 2 вида животных, в последующих долгосрочных исследованиях токсичности может быть достаточным использование только 1 вида животных (например, если в краткосрочных исследованиях профиль токсичности у 2 видов животных сопоставим).
Результаты исследований токсичности, проведенных на нерелевантных видах животных, могут вводить в заблуждение, поэтому их проведение не рекомендуется. Если релевантных видов животных не существует, можно использовать соответствующие виды трансгенных животных, у которых экспрессируются рецепторы, сходные по структуре с рецепторами человека (гуманизированные белки-рецепторы), или использовать гомологичные белки. Если взаимодействие между препаратом и рецептором человека оказывает сходные физиологические эффекты, сравнимые с теми, что ожидаются у человека, информация, полученная в ходе исследований на модели трансгенных животных, у которых экспрессируется рецептор человека, является наиболее адекватной. Полезная информация может быть получена и с использованием гомологичных белков. При этом следует учитывать, что производственный процесс, профиль примесей (контаминантов), фармакокинетика и точные фармакологические механизмы действия для гомологичной формы и исследуемого лекарственного препарата, предназначенного для клинического использования, могут различаться. Если использование трансгенных моделей и гомологичных белков невозможно, целесообразно оценить некоторые аспекты потенциальной токсичности в рамках ограниченных исследований токсичности на одном виде животных. Например, могут быть проведены исследования токсичности при многократном введении продолжительностью 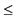 14 суток, которые включают оценку показателей функционирования наиболее важных систем организма (например, сердечно-сосудистой и дыхательной систем).
14 суток, которые включают оценку показателей функционирования наиболее важных систем организма (например, сердечно-сосудистой и дыхательной систем).
Следует учитывать прогресс, достигнутый в разработке животных моделей заболеваний, которые считаются сходными с заболеваниями человека. К ним относят животные модели в результате спонтанного или индуцированного развития заболеваний, нокаут гена (генов), а также трансгенные животные. Использование таких моделей может обеспечить проведение дальнейших исследований, которые ограничиваются не только изучением фармакологического действия препарата, его фармакокинетики и установлением диапазона доз. Модели могут быть использованы также и для исследования безопасности (например, для оценки перспективы нежелательного стимулирования прогрессирования заболевания). В некоторых случаях исследования, проведенные на животных моделях заболевания, могут использоваться в качестве подходящей альтернативы исследованиям токсичности на обычных животных (как это указано в пояснении 1 к настоящей главе). При этом необходимо представить научное обоснование использования животных моделей заболевания для исследования безопасности.
3.4. Число и пол животных
Число животных, используемых в расчете на одну дозу препарата, должно быть достаточным для оценки токсического действия препарата. Небольшой размер выборки животных может привести к тому, что признаки токсичности не будут обнаружены вследствие небольшой частоты их проявления, независимо от их тяжести. Ограничения, которые обусловлены недостатком выборки и часто наблюдаются в исследованиях на нечеловекообразных приматах (НЧП), могут быть частично компенсированы повышением частоты и продолжительности мониторинга за состоянием животных. Как правило, в экспериментах необходимо использовать животных обоих полов либо должно быть представлено обоснование в случае отсутствия таких данных.
3.5. Способ введения и выбор дозы
Способ и частота введений лекарственного препарата экспериментальным животным должны быть максимально приближены к условиям предполагаемым для клинического применения. Следует учитывать фармакокинетические характеристики исследуемого лекарственного препарата и его биодоступность у используемых видов животных. Кроме того, необходимо учитывать объем препарата, который можно вводить экспериментальным животным в соответствии с принципами безопасности и гуманности. Например, частота введения препарата лабораторным животным может быть увеличена по сравнению с предлагаемым режимом введения препарата субъектам клинических исследований. Причиной такого увеличения может быть необходимость компенсировать более высокий клиренс или низкую растворимость действующего вещества исследуемого препарата. В данном случае необходимо определить соотношение величины экспозиции препарата в крови подопытного животного и экспозиции препарата у человека в клинических условиях. Также следует учитывать зависимость эффекта от объема вводимого препарата, концентрации действующего вещества, состава и места введения. В некоторых случаях допускается изменение способа введения по сравнению с тем, который предполагается использовать в клинических исследованиях. Причинами такого изменения могут быть низкая биодоступность и ограничения, обусловленные способом введения или размером (физиологическими особенностями) используемых видов животных.
В целях получения сведений о зависимости "доза - эффект", включая токсическую дозу и высшую нетоксическую дозу (ВНТД, no observed adverse effect level (NOAEL)), необходимо выбрать исследуемые дозы. Установить максимальную дозу некоторых классов лекарственных препаратов с низкой или ничтожной токсичностью невозможно. В таких случаях необходимо представить научное обоснование стратегии выбора доз и предполагаемой кратности превышения экспозиции у человека. В целях обоснования выбора высоких доз необходимо проанализировать ожидаемые фармакологические (физиологические) эффекты, доступность подходящего исследуемого материала и предполагаемое клиническое применение. Если лекарственный препарат обладает низкой аффинностью или активностью в отношении клеток выбранного вида животных по сравнению с клетками человека, может потребоваться исследование более высоких доз. Границы безопасности диапазона доз у человека, подлежащие определению, могут варьироваться в зависимости от класса лекарственных препаратов, полученных биотехнологическим путем, и показаний к применению.
3.6. Иммуногенность
Многие лекарственные препараты для медицинского применения, полученные с использованием биотехнологических методов, являются иммуногенными у животных. Следовательно, определение концентрации антител, обусловленных введением препарата, необходимо осуществлять при проведении исследований токсичности при многократном введении (в соответствии с требованиями главы 5.4 настоящих Правил). Такие исследования необходимы для правильной интерпретации результатов токсикологических исследований. Необходимо охарактеризовать гуморальный иммунный ответ на введение исследуемого лекарственного препарата (например, титр антител, количество животных, у которых зарегистрирована выработка антител, свойства индуцированных антител (нейтрализующая активность или отсутствие такой активности)). Следует установить корреляцию между выработкой антител и любыми изменениями фармакологических и (или) токсических свойств исследуемого препарата на момент обнаружения антител. В частности, при интерпретации результатов исследования необходимо учитывать влияние образования антител на фармакокинетические (фармакодинамические) параметры препарата, частоту развития и (или) тяжесть проявления нежелательных явлений, активацию комплемента, появление новых токсических эффектов. Необходимо также оценить возможность появления патологических изменений, обусловленных образованием и отложением иммунных комплексов в тканях.
Выявление антител не должно служить единственным основанием для досрочного прекращения доклинических исследований безопасности или изменения их продолжительности, за исключением случаев, когда развитие иммунного ответа является причиной нейтрализации фармакологического и (или) токсического эффектов, оказываемых биологическим лекарственным препаратом, у значительной части животных. В большинстве случаев иммунный ответ на биотехнологический лекарственный препарат у животных (как и у человека) бывает вариабельным. Если все эти вопросы не влияют на интерпретацию данных, полученных в ходе исследования безопасности, то можно не придавать серьезного значения развитию гуморальной реакции иммунитета у экспериментальных животных.
Индукция образования антител у животных не позволяет прогнозировать развитие иммунного ответа при клиническом применении препарата. У человека могут формироваться циркулирующие антитела к гуманизированным белкам, причем терапевтический эффект в присутствии таких антител часто сохраняется. У человека частота развития тяжелой анафилактической реакции в ответ на введение рекомбинантных белков невелика. В связи с этим результаты тестов на анафилактические реакции у морских свинок, которые, как правило, положительны для белковых препаратов, обычно не позволяют спрогнозировать реакцию у человека. В связи с этим, такие исследования считаются малоинформативными для стандартного изучения данного типа препаратов.
4. Частные требования
4.1. Фармакологическая безопасность
Необходимо исследовать потенциальный риск развития нежелательных реакций, обусловленных нежелательной фармакологической активностью препарата, на соответствующих моделях животных. При необходимости следует включить контроль такой активности в программу исследований токсичности и (или) клинических исследований. В ходе исследований фармакологической безопасности обычно определяют функциональные индексы потенциальной токсичности препарата. Указанные функциональные индексы могут быть изучены в рамках отдельных исследований или включены в токсикологические исследования. Целью исследований фармакологической безопасности должно быть выявление влияния лекарственного препарата на жизненные функции основных физиологических систем организма (в том числе на сердечно-сосудистую, респираторную, выделительную, центральную нервную системы). Кроме того, вместо животных в ходе исследований могут быть использованы изолированные органы или другие тест-системы без привлечения интактных животных. Подобные исследования позволяют объяснить возникновение органоспецифической токсичности, однако следует с осторожностью подходить к интерпретации этих результатов, учитывая показания и особенности применения препарата у человека.
4.2. Оценка экспозиции
4.2.1. Фармакокинетика и токсикокинетика.
Единой схемы проведения фармакокинетических исследований биотехнологических лекарственных препаратов не имеется. Информативными являются фармакокинетические исследования при однократном и многократном введении лекарственного препарата, токсикокинетические исследования, исследования распределения в тканях на релевантных видах животных. В то же время стандартные исследования, направленные на оценку материального баланса введенного и элиминированного препарата, являются малоинформативными. Различия в фармакокинетических параметрах исследуемого препарата у разных видов животных могут иметь существенное значение для прогнозирования результатов при исследовании на животных и оценки кривой зависимости "доза - эффект" при исследовании токсичности. Изменения фармакокинетического профиля вследствие иммуноопосредованных механизмов элиминации могут повлиять на кинетические профили и интерпретацию результатов исследований токсичности. Для некоторых лекарственных препаратов (например, для цитокинов) может быть характерна значительная задержка в проявлении фармакодинамических эффектов относительно фармакокинетических характеристик. Кроме того, возможно пролонгирование фармакодинамических эффектов по сравнению с содержанием действующего вещества в плазме.
Фармакокинетические исследования следует по мере возможности проводить с использованием лекарственных препаратов, являющихся репрезентативными по отношению к препаратам, предназначенным для токсикологических исследований и клинического применения. Способ и частота их дозирования должны быть максимально приближены к тем, которые соответствуют планируемым клиническим исследованиям. Профиль абсорбции может зависеть от состава, концентрации, области введения и (или) объема вводимого препарата. По возможности во время проведения исследований токсичности необходимо осуществлять мониторинг системной экспозиции.
При использовании белков с радиоактивной меткой важно доказать, что материал с радиометкой, используемый для исследования, сохраняет активность и биологические свойства, эквивалентные таковым у немеченного вещества. Величину радиоактивности в тканях и (или) данные ауторадиографии при использовании белков с радиоактивной меткой бывает сложно интерпретировать ввиду быстрого метаболизма белка in vivo или нестабильности связи радиоактивной метки с белком. Результаты исследований, в которых радиоактивная метка вводится в специфичные аминокислоты, необходимо интерпретировать с осторожностью. Следует учитывать возможность высвобождения радиоактивной метки, встроенной в специфические аминокислоты, и ее включение в аминокислоты, которые участвуют в синтезе белков и пептидов, не имеющих отношения к исследуемому лекарственному препарату.
Необходимо располагать начальной информацией об особенностях абсорбции, распределения и клиренса исследуемого лекарственного препарата у релевантных видов животных до начала клинического исследования с целью прогнозирования широты терапевтического действия препарата (безопасного диапазона доз) с учетом экспозиции и дозы.
4.2.2. Аналитические методы.
Использование одного или нескольких методов количественного определения следует рассматривать в индивидуальном порядке, при этом необходимо представить научное обоснование. Как правило, достаточно одной валидированной методики. Например, достоверную информацию можно получить методом количественного определения радиоактивности в преципитате (белок с радиоактивной меткой, осажденный трихлоруксусной кислотой). В то же время более предпочтительным является специфичный метод количественного определения анализируемого вещества. Оптимально использовать одни и те же методы количественного определения как в исследованиях на животных, так и в исследованиях с участием человека. Необходимо определить потенциальное влияние связывающих белков плазмы и (или) антител в плазме (сыворотке) крови на результаты количественного определения исследуемого вещества.
4.2.3. Метаболизм.
Следствием метаболизма биотехнологических лекарственных препаратов является их деградация до небольших пептидов и отдельных аминокислот. Пути метаболизма таких препаратов в целом изучены. Следовательно, нет необходимости в проведении классических исследований биотрансформации, проводимых в отношении лекарственных препаратов, действующие вещества которых получены путем химического синтеза.
Для понимания природы фармакодинамического эффекта необходимы сведения о поведении биологических лекарственных препаратов в биологических жидкостях (например, в плазме, сыворотке, цереброспинальной жидкости), а также о возможном влиянии связывания с белками.
4.3. Исследования токсичности при однократном введении
По результатам исследований токсичности при однократном введении можно получить полезную информацию для описания зависимости системной и (или) местной токсичности от дозы. Эти данные могут быть использованы для выбора доз при исследовании токсичности при многократном введении. Информация о зависимости "доза - эффект" может быть получена при проведении исследований токсичности при однократном введении, которые являются частью программы исследований фармакологической активности или эффективности с использованием животных моделей. Следует рассмотреть возможность включения в данную программу фармакологических исследований оценку показателей безопасности лекарственного препарата.
4.4. Исследования токсичности при многократном введении
В подразделе 3.3 настоящей главы приведены принципы выбора видов животных для проведения исследований с многократным введением. Способ введения и режим дозирования (например, ежедневное введение или периодическое дозирование) должны быть максимально приближены к предлагаемому клиническому применению или воздействию. По возможности данные исследования должны включать изучение токсикокинетики.
Как правило, дизайн исследования должен предусматривать период последующего наблюдения после прекращения введения препарата. Это необходимо для того, чтобы выявить обратимость эффекта, потенциальное усиление фармакологических и (или) токсических эффектов или потенциально отсроченные токсические эффекты. Для биотехнологических лекарственных препаратов, которые оказывают длительные фармакологические и (или) токсические эффекты, группы животных в период восстановления должны находиться под наблюдением до тех пор, пока не будет продемонстрирована обратимость эффектов. Продолжительность исследований токсичности при многократном введении должна зависеть от предполагаемой длительности клинического применения и показаний к применению. Для большинства биотехнологических лекарственных препаратов длительность введения животным, как правило, составляет 1 - 3 месяца. Для препаратов, предназначенных для кратковременного применения (например, не более 7 дней), а также для лечения острых, угрожающих жизни заболеваний, достаточным считается проведение исследований токсичности при многократном введении в течение 2 недель. Как правило, этого времени достаточно для обоснования возможности проведения клинических исследований и регистрации лекарственного препарата. Для биотехнологических лекарственных препаратов, которые предназначены для лечения хронических заболеваний и применяются длительно, достаточными являются исследования продолжительностью 6 месяцев. Однако в некоторых случаях возможно проведение более коротких или более длительных исследований для включения их результатов в регистрационное досье. Продолжительность долгосрочных исследований токсичности биологических лекарственных препаратов, предназначенных для длительного применения, должна быть научно обоснована.
4.5. Исследования иммунотоксичности
Одним из аспектов оценки иммунотоксичности является оценка потенциальной иммуногенности (в соответствии с подразделом 3.6 настоящей главы). Многие биотехнологические лекарственные препараты предназначены для стимуляции или подавления иммунной системы. Следовательно, они могут оказывать влияние не только на гуморальный, но и на клеточный иммунитет. Воспалительные реакции в месте инъекции могут свидетельствовать о стимуляции иммунной системы организма в ответ на введение препарата. Однако необходимо учитывать, что обычная травма во время инъекции и (или) токсические эффекты, обусловленные веществами, входящими в состав лекарственного препарата, могут привести к токсическим изменениям в области инъекции. Кроме того, возможно изменение экспрессии антигенов на мембране клеток-мишеней, что может иметь значение для развития аутоиммунного ответа. Для уточнения подобных вопросов в схемы изучения иммунотоксических свойств препарата может быть включено проведение скрининговых исследований с последующей оценкой механизма действия лекарственного препарата. Однако применение стандартного пошагового подхода или проведение стандартного набора тестов в отношении биотехнологических лекарственных препаратов не рекомендовано.
4.6. Исследование репродуктивной
и онтогенетической токсичности
Необходимость проведения исследований репродуктивной и онтогенетической токсичности зависит от свойств лекарственного препарата, показаний к применению и целевой популяции пациентов (как это указано в пояснении 2 к настоящей главе). Дизайн исследования и режим дозирования могут быть изменены в зависимости от факторов, обусловленных видоспецифичностью, иммуногенностью, биологической активностью и (или) длительным периодом полувыведения конкретного лекарственного препарата. Например, подозрения на наличие потенциальной онтогенетической иммунотоксичности, которые могут иметь место в отношении определенных моноклональных антител с длительным иммунологическим эффектом, могут быть учтены путем изменения дизайна исследования, для того, чтобы оценить иммунологический статус новорожденного.
4.7. Исследования генотоксичности
Диапазон и виды исследований генотоксичности, стандартно проводимые в отношении лекарственных препаратов, действующие вещества которых получены путем химического синтеза, не применимы в отношении биотехнологических лекарственных препаратов, поэтому проведение таких исследований не требуется. Кроме того, введение большого количества веществ пептидной (белковой) природы может привести к получению результатов, не поддающихся интерпретации. Эти вещества, вероятно, не взаимодействуют напрямую с ДНК или другим хромосомным материалом (как это указано в пояснении 3 к настоящей главе).
Если имеются основания предполагать наличие таких взаимодействий (например, ввиду присутствия органической молекулы-линкера в препарате на основе конъюгированного белка), необходимо проведение исследований с использованием доступных и подходящих систем, а также с включением новых тест-систем. Использование стандартных исследований генотоксичности для оценки генотоксического потенциала производственных примесей неприемлемо. При проведении таких исследований с этой целью требуется соответствующее обоснование.
4.8. Исследования канцерогенности
Как правило, для биотехнологических лекарственных препаратов стандартные тесты на канцерогенность с использованием биологических систем неприемлемы. В то же время может быть необходима оценка канцерогенности с использованием методов, специфичных для определенного препарата. Это зависит от предполагаемой продолжительности клинического применения препарата, популяции пациентов и (или) его биологической активности (например, факторы роста, иммунодепрессанты и др.). В случае, если существуют опасения относительно канцерогенного потенциала препарата, могут быть использованы разные подходы для оценки такого риска.
Некоторые препараты могут обладать потенциальной способностью вызывать пролиферацию трансформированных клеток и клональную экспансию (экспансию клона), что может привести к развитию новообразований. В этом случае необходимо провести оценку препарата с учетом экспрессии рецепторов на различных злокачественных и нормальных клетках человека, которые потенциально значимы в отношении исследуемой популяции пациентов. Необходимо определить способность препарата стимулировать рост нормальных или злокачественных клеток, экспрессирующих соответствующие рецепторы. Если в результате исследований in vitro получены данные, свидетельствующие о возможном канцерогенном потенциале препарата, необходимо провести дальнейшие исследования на подходящих животных моделях. Ценная информация может быть получена при включении в долгосрочные исследования токсичности с многократным введением оценки чувствительных показателей пролиферации клеток у экспериментальных животных.
Если препарат биологически активен и не вызывает развитие иммунного ответа у грызунов, однако информации, полученной на основании результатов других исследований, недостаточно для оценки канцерогенного потенциала препарата, использование одного вида грызунов является оправданным. Особенно внимательно необходимо подойти к выбору дозы. Научно обоснованный подход к определению подходящих доз должен быть основан на комплексном анализе фармакокинетических и фармакодинамических конечных точек с учетом сравнительных характеристик рецепторов и предполагаемой экспозиции у человека. Выбранная дозировка должна быть обоснована.
4.9. Исследования местной переносимости
Необходимо провести оценку местной переносимости. При этом необходимо использовать лекарственный препарат, имеющий состав, который в дальнейшем будет представлен к регистрации. Однако при достаточном обосновании допускается использование препарата, имеющего репрезентативный состав. В отдельных случаях потенциальные нежелательные реакции лекарственного препарата могут быть оценены в рамках исследований токсичности при однократном или многократном введении. Таким образом, исключается необходимость проведения отдельных исследований по оценке местной переносимости.
Пояснения
1. Использование животных моделей заболеваний может быть целесообразным при определении показателей токсичности, выборе клинических показаний и определении подходящей лекарственной формы, пути введения и режима дозирования. Следует учесть, что при использовании таких моделей заболевания часто имеется слишком мало ретроспективных данных для использования в качестве контроля при оценке результатов исследований. Следовательно, для оптимизации дизайна исследований критически важно параллельное получение контрольных и исходных данных.
2. Возможна ситуация, когда в опубликованных данных научной литературы имеется значительный объем информации, касающейся потенциального воздействия на репродуктивную функцию и (или) эмбриогенез определенных групп биотехнологических препаратов (например, интерферонов), которая была получена и может быть подтверждена только в исследованиях на единственном релевантном виде животных - нечеловекообразных приматах. В таких случаях исключается необходимость проведения формальных исследований репродуктивной (онтогенетической) токсичности, если установлено, что структура и физико-химическая характеристика действующего вещества сопоставимы, поскольку родственные молекулы будут вызывать сходные биологические эффекты. В каждом конкретном случае должно быть приведено научное обоснование оценки потенциального влияния препарата на репродуктивную функцию и развитие потомства.
3. В отношении некоторых биотехнологических лекарственных препаратов существуют опасения в отношении аккумуляции спонтанно мутировавших клеток (например, за счет избирательного усиления пролиферации), что может способствовать канцерогенному эффекту таких лекарственных препаратов. Стандартный набор исследований генотоксичности не подходит для выявления подобных ситуаций. Для изучения данного аспекта производителю следует разработать и проанализировать альтернативные модели in vitro или in vivo.
Глава 5.4. Доклиническая оценка безопасности лекарственных
препаратов, полученных с использованием биотехнологических
методов (дополнительные требования)
1. Введение
1.1. Цели дополнительных требований
Цель настоящих требований дополнить и разъяснить отдельные положения главы 5.3 настоящих Правил:
выбор видов животных;
дизайн исследования;
иммуногенность;
изучение репродуктивной и онтогенетической токсичности;
оценку канцерогенного потенциала.
Настоящие Правила должны способствовать своевременному проведению клинических исследований, сокращению использования животных при доклинических исследованиях в соответствии с принципом "трех R" (сократить/оптимизировать/заменить "reduce/refine/replace") и сокращению использования других ресурсов при разработке лекарственных препаратов. Необходимо рассмотреть возможность использования соответствующих альтернативных методов оценки безопасности in vitro, хотя данный вопрос не затрагивается в настоящих Правилах. Эти методы следует применять всем уполномоченным органам государств-членов в сфере обращения лекарственных средств, что позволит заменить для данной группы лекарственных препаратов стандартные методы изучения.
Настоящая глава Правил позволяет обеспечить безопасную, этичную разработку и доступность новых биотехнологических лекарственных препаратов.
1.2. Справочная информация
Требования, представленные в настоящей главе, обеспечивают гармонизацию доклинических исследований безопасности биотехнологических лекарственных препаратов, проводимых на различных этапах их клинической разработки в разных государствах-членах.
1.3. Область применения
Настоящая глава не вносит изменений в область применения главы 5.3 настоящих Правил. Для лекарственных препаратов, полученных с использованием биотехнологических методов, которые предназначены для использования в онкологии, следует использовать указания, изложенные в соответствующих научных руководствах и актах, входящих в право Союза.
2. Выбор видов животных
2.1. Общие принципы
При оценке релевантности вида животных, выбранных для проведения исследований, следует принять во внимание несколько факторов. На первом этапе следует провести сравнение последовательностей молекул-мишеней с целью выявления гомологии между видами животных. На следующем этапе должна проводиться в тестах in vitro качественная и количественная межвидовая сравнительная оценка аффинности связывания действующего белкового вещества препарата с мишенью, распределения (занятости) рецепторов (лигандов) и кинетических характеристик такого связывания.
Также следует выполнить оценку функциональной активности. Функциональная активность может быть продемонстрирована в исследованиях с использованием видоспецифичных клеточных систем и (или) в рамках фармакологических и токсикологических исследований in vivo. Доказательством функциональной активности, которое можно использовать для обоснования выбора данного вида животных, может быть модуляция биологической реакции или показателей фармакодинамического маркера.
При изучении различий между видами по связыванию лекарственного препарата с мишенью и его функциональной активности в контексте предполагаемого режима дозирования необходимо представить обоснование, что выбранная модель способна выявлять потенциально нежелательные последствия, связанные с модуляцией антигена-мишени. В некоторых случаях у обычно используемых в доклинических исследованиях видов здоровых животных, уровень экспрессии антигенов-мишеней (например, провоспалительные цитокины или опухолевые антигены) чрезвычайно низкий и для обоснования выбора вида животных достаточно определить аффинность связывания и активность лекарственного препарата на клеточных системах.
При выборе вида животных оценка перекрестной тканевой реактивности на тканях животных имеет ограниченное значение (как это указано в пояснении 1). В отдельных случаях (если описанные выше подходы не могут быть использованы для поиска видов животных, релевантных по фармакологическим параметрам) при выборе видов животных для токсикологических исследований могут быть использованы результаты исследований перекрестной тканевой реактивности (ПТР или TCR). При этом необходимо провести сравнение профилей связывания с тканями у человека и животных, с которыми предполагается связывание мишени.
Как указано в главе 5.4 настоящих Правил, при отсутствии релевантных видов животных по той причине, что биологический лекарственный препарат не взаимодействует с мишенями-ортологами ни у одного из исследуемых видов животных, допускается возможность использования гомологичных молекул или трансгенных моделей.
Для моноклональных антител и других препаратов на основе антител, направленных против чужеродных мишеней (бактериальные, вирусные антигены и др.), может быть рассмотрена возможность проведения краткосрочного исследования безопасности (в соответствии с положениями главы 5.3 настоящих Правил) с использованием одного вида животных (выбор такого вида животных должен быть обоснован). При этом нет необходимости в проведении дополнительных исследований токсичности, в том числе репродуктивной токсичности. В качестве альтернативного варианта можно включить оценку безопасности, если используется животная модель заболевания для проверки и подтверждения механизма действия лекарственного препарата. В этом случае целью оценки безопасности будет получение информации об опосредованных мишенью потенциальных угрозах безопасности. Если это сделать невозможно, следует разработать подходящие стратегии для снижения риска при проведении клинических исследований.
Выбор вида животных при исследованиях конъюгатов антител с лекарственными препаратами или токсинами (ADC), в которых используется новый токсин (токсикант), проводится в соответствии с теми же основными принципами, которые применяются и для неконъюгированных антител (как это указано в пояснении 2).
2.2. Выбор 1 или 2 видов животных
Если 2 вида животных являются фармакологически релевантными (один из отряда грызунов и другой, не относящийся к грызунам), в краткосрочных исследованиях общей токсичности (продолжительностью до 1 месяца) следует использовать оба вида животных. Если результаты обоих токсикологических исследований сходны или могут быть объяснены известным механизмом действия препарата, долгосрочные исследования общей токсичности достаточно провести только на одном виде животных. При этом следует использовать грызунов, если только для альтернативного подхода нет научного обоснования. Исследования на двух видах, не относящихся к грызунам, считаются неприемлемыми.
Использование одного вида животных для всех исследований общей токсичности обосновано только в том случае, если исследуемый лекарственный препарат, который предполагается использовать в клинической практике, фармакологически активен только у этого вида. При этом исследования на другом виде животных с использованием гомологичного продукта не могут представить дополнительные данные по оценке риска и поэтому их не следует проводить.
2.3. Использование гомологичных белков
Использование гомологичных белков является одним из альтернативных подходов, описанных в подразделе 3.3 главы 5.3 настоящих Правил. Исследования, в которых используются гомологичные белки, проводятся для выявления опасностей и оценки потенциала развития нежелательных реакций, обусловленных усиленным фармакологическим действием препарата. В то же время такие исследования, как правило, не информативны для количественной оценки риска. Именно поэтому для выявления риска следует проводить исследования безопасности с одной контрольной группой и одной группой, получающей препарат. При этом необходимо представить научное обоснование дизайна исследования и выбранной дозы (например, максимальная фармакологическая доза).
3. Дизайн исследования
3.1. Выбор дозы и применение принципов
фармакокинетики и фармакодинамики
Токсичность большинства биологических лекарственных препаратов связана с направленным механизмом их действия. В связи с этим относительно высокие дозы могут вызвать нежелательные реакции, которые являются следствием чрезмерного фармакологического действия препарата.
При выборе доз необходимо представить научное обоснование с учетом характеристик зависимости "доза - эффект". Выбору высоких доз способствует использование принципов, применяемых при анализе данных фармакокинетики и фармакодинамики (например, выявление простой зависимости "экспозиция - эффект" или более сложные подходы, основанные на моделировании и симуляции). При этом возможны следующие варианты выявления доз:
доза, которая обеспечивает максимальный ожидаемый фармакологический эффект у выбранного вида животных, используемых в доклинических исследованиях;
доза, которая в 10 раз превышает максимальную дозу, предполагаемую для исследования в клинических условиях.
Для группы с высокой дозой в доклинических токсикологических исследованиях необходимо выбрать наибольшую из этих 2 доз. В противном случае необходимо представить обоснование использования более низкой дозы (например, максимально допустимой дозы).
Если нет доступных in vivo (ex vivo) фармакодинамических конечных точек, выбор высокой дозы может быть основан на данных фармакокинетики, доступных данных по связыванию лекарственного препарата с рецептором в исследованиях in vitro и (или) фармакологических данных. При выборе верхней границы экспозиции в клинических условиях необходимо провести коррекцию с учетом степени связывания лекарственного препарата с мишенью и фармакологической активностью in vitro между человеком и видами животных, которые используются в доклинических исследованиях. В частности, значительное относительное различие в аффинности и (или) активности в условиях in vitro может свидетельствовать о целесообразности изучения высоких доз в рамках доклинических исследований. Если при использовании данного подхода не удается обнаружить токсичность выбранных доз, то маловероятно, что дополнительные токсикологические исследования более высоких доз, кратных дозам, применяемым у человека, приведут к получению необходимой значимой информации.
3.2. Продолжительность исследований
Для препаратов, которые предполагается использовать в течение длительного времени, считается достаточным проведение исследований токсичности при многократном введении на грызунах или негрызунах продолжительностью 6 месяцев. В таких исследованиях должны быть использованы высокие дозы, выбранные в соответствии с принципами, изложенными выше (в соответствии с требованиями подраздела 3.1 настоящей главы). Как правило, исследования с большей продолжительностью не позволяют получить дополнительной информаций, которая повлияла бы на клиническую разработку.
Принципы определения продолжительности токсикологических исследований для биотехнологических лекарственных препаратов, разрабатываемых для длительного применения пациентами с распространенным раком, должны соответствовать актам, входящим в право Союза и научным руководствам в области онкологии.
3.3. Восстановление
Необходимо оценить восстановление животных после фармакологических и токсикологических эффектов, имеющих потенциальное значение для развития нежелательных реакций у человека и возникающих в клинически значимых дозах. Необходимая информация может быть получена при установлении обратимости (необратимости) конкретного наблюдаемого эффекта или путем включения периода без дозирования как минимум в одно исследование и как минимум в одной дозе, что должно быть обосновано спонсором. Цель периода без введения исследуемого препарата заключается в установлении обратимости обнаруженных эффектов, а не в оценке проявлений отсроченной (поздней) токсичности. Демонстрация полного восстановления не требуется. Включение периода восстановления только с целью оценки иммуногенного потенциала исследуемого препарата не требуется.
3.4. Поисковые клинические исследования
К доклиническим исследованиям безопасности биологических лекарственных препаратов в целях проведения клинических исследований и регистрации могут быть применимы гибкие подходы, описанные актах, входящих право Союза и научных руководствах. Разработчикам лекарственного препарата следует обсудить и согласовать эти подходы с уполномоченными органами государств-членов.
4. Иммуногенность
Оценка иммуногенности проводится для того, чтобы обеспечить правильную интерпретацию результатов исследований и разработку дизайна дальнейших исследований. Такой анализ в ходе доклинических исследований на животных неинформативен в плане прогнозирования потенциальной иммуногенности человеческих или гуманизированных белков у человека.
Определение концентрации антител, вырабатываемых в ответ на биотехнологический лекарственный препарат (ADA) в рамках доклинических исследований, должно проводиться в следующих случаях:
при наличии доказательства изменения фармакодинамической активности;
при получении данных о неожиданном изменении экспозиции в отсутствие фармакодинамических маркеров;
при выявлении иммуноопосредованных реакций в ответ на введение препарата (болезнь иммунных комплексов, васкулиты, анафилактические реакции и т.д.).
Поскольку до завершения исследования трудно определить, будет ли необходимо проведение такого анализа, во многих случаях целесообразно получить соответствующие образцы в ходе исследования. В дальнейшем эти образцы могут быть проанализированы и в случае необходимости использованы для интерпретации результатов исследования. Если в ходе исследования обнаружены антитела к лекарственному препарату, потребуется оценка их влияния на интерпретацию результатов исследования (дополнительные требования к оценке влияния иммуногенности приведены в подразделе 3.6 главы 5.3 настоящих Правил).
При выявлении антител и одновременном отсутствии фармакодинамических маркеров, позволяющих продемонстрировать устойчивую активность таких антител в условиях токсикологических исследований in vivo, необходима оценка нейтрализующей активности антител. Нейтрализующую активность антител можно оценить косвенно с помощью биологических методов ex vivo, путем соответствующей комбинации различных видов анализа показателей фармакокинетики (фармакодинамики) или непосредственно путем использования специальных методов, определяющих нейтрализующую способность антител.
5. Репродуктивная и онтогенетическая токсичность
5.1. Требования общего характера
Исследования репродуктивной токсичности препарата следует проводить в соответствии с актами, входящими в право Союза и научными руководствами. Исходя из знаний о видоспецифичности, природе исследуемого лекарственного препарата и механизме его действия, иммуногенности и (или) фармакокинетических свойствах, а также об экспозиции в период эмбриофетального развития, отдельный дизайн исследования и режим дозирования могут варьироваться.
В целом предпочтительной является оценка репродуктивной токсичности на релевантных видах животных с использованием лекарственного препарата, который предполагается применять в клинических исследованиях. Оценка репродуктивной токсичности должна проводиться на видах животных, которые подходят по фармакологическим параметрам. Если препарат, который предполагается использовать в клинике, проявляет свою фармакологическую активность у грызунов и кроликов, то оба вида этих животных должны использоваться в исследованиях эмбриофетальной токсичности (ЭФТ (EFD)). Исключением являются случаи, когда для одного вида животных установлена эмбриофетальная летальность или тератогенное действие.
Исследования онтогенетической токсичности на нечеловекообразных приматах (НЧП) допускается проводить только в случае, если они являются единственным релевантным видом животных.
Если исследуемый препарат является фармакологически активным только у НЧП, решение принимается в пользу проведения исследований препарата на данном виде животных. Однако вместо приматов изучение препарата-кандидата для клинических исследований может быть проведено с использованием альтернативных видов животных, если представлено соответствующее научное обоснование для выбора альтернативной модели.
Если релевантные виды животных, на которых могут быть проведены клинические исследования препарата-кандидата, отсутствуют, возможны следующие решения:
допускается использование трансгенных мышей, которые экспрессируют мишень человека, или использование гомологичных белков соответствующих видов животных, экспрессирующих ортолог человеческого белка-мишени. Для этого необходимо, чтобы относительно используемой модели было получено достаточно информации (например, данные предшествующих исследований) (как это указано в пояснении 1 к главе 5.3 настоящих Правил). Для лекарственных препаратов, которые направлены против чужеродных мишеней (например, бактериальных или вирусных), как правило, нет необходимости проводить исследования репродуктивной токсичности (в соответствии с требованиями подраздел 2.1 настоящей главы).
Если существуют весомые доказательства того, что препарат оказывает нежелательное влияние на фертильность или течение беременности (например, механизм действия, фенотипические данные, полученные на генетически модифицированных животных, сведения об эффектах препаратов, относящихся к данной группе), они могут представить достаточную информацию о существовании риска для репродуктивной системы. В этом случае при соответствующих условиях проведение дополнительных доклинических исследований безопасности может не потребоваться.
5.2. Фертильность
Оценка фертильности при исследовании препаратов, для которых мыши и крысы являются фармакологически релевантными видами, может проводиться на одном виде грызунов (в соответствии с требованиями актов, входящих в право Союза, и научными руководствами по проведению исследований репродуктивной токсичности). Дизайн исследований может быть адаптирован для других видов животных, если они являются фармакологически релевантными. Кроме того, дизайн исследования должен быть соответствующим образом дополнен для того, чтобы, например, в результате исследования была охарактеризована природа лекарственного препарата и проведена оценка его иммуногенного потенциала.
При работе с НЧП проведение исследований со спариванием является нецелесообразным. В то же время, если НЧП являются единственным релевантным видом животных, может быть проведена оценка потенциального влияния препарата на фертильность самцов и самок. Для этого необходимо оценить влияние препарата на репродуктивную систему животных (например, массу органов и гистопатологические изменения) в рамках исследований токсичности с многократным введением длительностью не менее 3 месяцев. Для исследований необходимо использовать половозрелых НЧП. Если на основании данных о фармакологической активности или результатов предыдущих исследований существуют опасения относительно токсичности лекарственного препарата, может быть проведена оценка специальных параметров в ходе исследований токсичности многократного введения препарата. К таким параметрам относятся периодичность менструального цикла, число сперматозоидов, морфология и (или) подвижность сперматозоидов, содержание половых гормонов у самцов и самок.
Если НЧП являются единственным релевантным видом животных и при этом существуют теоретические предпосылки относительно потенциального влияния препарата на оплодотворение (имплантацию), причиной которого является его фармакологическая активность, следует проверить такие теоретические предпосылки экспериментально. В случае наличия данных предпосылок единственной практической возможностью оценить потенциальные эффекты на оплодотворение и имплантацию является использование гомологичных белков или трансгенных моделей. В то же время не рекомендуется разрабатывать гомологичные белки или трансгенные модели с единственной целью проведения исследований репродуктивной функции у грызунов. При отсутствии доклинических данных минимизацию риска следует осуществлять путем выполнения соответствующих процедур в рамках клинических исследований, получения информированного согласия у субъектов клинического исследования и внесения соответствующих сведений в информацию о лекарственном препарате.
5.3. Эмбриофетальное развитие и пренатальное
(постнатальное) развитие потомства
При выборе дизайна и интерпретации результатов исследования по оценке эмбриофетальной токсичности и влияния отдаленных эффектов токсичности на развитие потомства необходимо учитывать потенциальные различия между биотехнологическими лекарственным препаратами в отношении их способности проникать через плацентарный барьер (как указано в пояснении 3 к настоящей главе).
В отношении изучения лекарственных препаратов, проявляющих свою фармакологическую активность только у НЧП, может быть использовано несколько дизайнов исследования с учетом предполагаемого клинического применения и ожидаемых фармакологических эффектов. Целесообразно проведение отдельных исследований на этапах, охватывающих эмбриофетальное и (или) пренатальное (постнатальное) развитие (ППНР (PPND)). Возможны и другие дизайны исследования при условии их обоснования, особенно при наличии некоторых опасений относительно того, что нежелательное влияние на эмбриофетальное развитие или смерть плода во время беременности могут быть вызваны самим механизмом действия исследуемого лекарственного препарата. В ряде случаев более целесообразно проведение одного тщательно спланированного исследования на НЧП, которое предусматривает введение лекарственного препарата самкам с 20-го дня беременности и до рождения потомства (усиленное ППНР (ePPND)), чем проведение отдельных исследований ЭФТ и (или) ППНР.
При проведении единственного исследования усиленного ППНР, которое предусматривает изучение влияния препарата на пренатальное (постнатальное) развитие плода и дизайн которого описан выше, включение группы животных, которым проводят кесарево сечение, является нецелесообразным, при этом необходимо проводить оценку исходов беременности, завершившихся естественными родами. В рамках указанного исследования необходимо оценить жизнеспособность потомства, внешние пороки развития, аномалии развития скелета (например, с помощью рентгенографии), а также морфологию внутренних органов и тканей при вскрытии животных. Проведение ультразвуковых исследований эффективно для контроля протекания беременности, однако такие исследования являются малоинформативными для обнаружения пороков развития потомства. Оценка возможных пороков развития осуществляется в период послеродового наблюдения. Введение исследуемого препарата матери в послеродовый период не рекомендуется, поскольку это может оказывать неблагоприятное влияние на материнскую заботу о потомстве. При необходимости, в исследование можно включить анализ других конечных точек (параметров, оцениваемых у потомства), если они имеют значение для оценки фармакологической активности препарата. Продолжительность постнатальной фазы исследований зависит от дополнительных конечных точек, которые выбирают в соответствии с механизмом действия исследуемого лекарственного препарата (как это указано в пояснении 4 к настоящей главе).
Исследования онтогенетической токсичности на НЧП позволяют выполнить только идентификацию рисков. Число животных в группе должно быть достаточным для достоверной интерпретации полученных данных (как это указано в пояснении 5 к настоящей главе).
При использовании других видов НЧП необходимо представить обоснование дизайна такого исследования. Перечисленные выше исследования онтогенетической токсичности, которые проводятся на НЧП, направлены только на выявление возможной угрозы, поэтому проведение таких исследований возможно с включением одной исследуемой группы (получающей одну дозу препарата) и одной контрольной группы. При этом необходимо представить научное обоснование выбора дозы. В качестве примера научного обоснования может быть рассмотрен опыт исследования моноклональных антител, способных связываться с растворимым антигеном-мишенью. При этом используются предлагаемые для клинического применения дозы препарата моноклональных антител, обеспечивающие насыщение его связывания с мишенью. Если такое насыщение связывания с мишенью может быть продемонстрировано на выбранном для исследования виде животных и применяемая доза обеспечивает экспозицию, которая не более чем в 10 раз превышает экспозицию достигаемую при клиническом применении, результаты исследования единственной дозы при наличии контрольной группы животных обеспечивают достаточную оценку угрозы исследуемого препарата на эмбриофетальное развитие.
5.4. Сроки проведения исследования
Если в клинические исследования включаются женщины с детородным потенциалом, до получения информации о возможном влиянии препарата на эмбриофетальное развитие, должны быть приняты соответствующие меры по ограничению клинического риска, например, даны рекомендации по использованию высокоэффективных методов контрацепции (в соответствии с актами, входящими в право Союза и регламентирующими выполнение доклинических исследований безопасности в целях дальнейшего проведения клинических исследований и регистрации лекарственных препаратов).
Для биотехнологических лекарственных препаратов, которые проявляют фармакологическую активность только у НЧП, доклинические исследования ЭФТ и усиленного ППНР могут проводиться параллельно с клиническими исследованиями фазы III, при условии применения мер предосторожности, достаточных для предотвращения наступления беременности у субъектов клинического исследования. В этом случае отчет о результатах проведенных исследований необходимо представить при подаче заявления о регистрации лекарственного препарата. Если спонсор не может обеспечить достаточные меры для предотвращения беременности у женщин, включенных в клинические исследования, необходимо до начала фазы III представить полный отчет об исследовании токсичности при ППНР, либо промежуточный отчет об исследовании токсичности в усиленном ППНР (как это указано в пояснении 6 к настоящей главе). Если препарат является фармакологически активным только у НЧП, и механизм его действия позволяет сделать теоретическое заключение относительно его влияния на эмбриофетальное развитие, представляемая субъектам клинического исследования информация о лекарственном препарате должна включать указание о возможном существовании таких последствий. При этом проведение исследования онтогенетической токсичности на НЧП не требуется. В инструкции по применению необходимо указать, что женщинам с детородным потенциалом следует избегать применения такого лекарственного препарата.
Если релевантными видами животных являются грызуны или кролики, информация о сроках проведения исследований репродуктивной токсичности должна соответствовать требованиям актов, входящих в право Союза, и регламентирующих выполнение доклинических исследований безопасности в целях дальнейшего проведения клинических исследований и регистрации лекарственных препаратов. Указанных требований следует придерживаться также в вопросах о сроках проведения исследований при оценке влияния на фертильность тех препаратов, для которых грызуны являются релевантными видами животных.
Для препаратов, соответствующих требованиям актов, входящих в право Союза по доклинической оценке безопасности противоопухолевых лекарственных препаратов и препаратов, предназначенных для лечения онкологических заболеваний, вопросы, связанные со сроками проведения исследований, должны также соответствовать положениям настоящей главы.
6. Канцерогенность
Необходимость проведения специфичных для конкретного биологического лекарственного препарата исследований канцерогенного потенциала следует определять исходя из популяции, у которой планируется клиническое применение препарата, а также длительностью его применения (как это указано в актах, входящих в право Союза и регламентирующих выполнение соответствующих доклинических исследований безопасности). Если такая оценка необходима, заявитель должен разработать программу исследований, которая позволила бы выявить потенциальную угрозу препарата.
Программа исследования должна основываться на анализе совокупности существующих данных, в том числе на обзоре соответствующих сведений, полученных из разного рода источников. Среди источников информации могут быть научные медицинские публикации (например, информация, полученная в результате исследования моделей заболевания у животных, трансгенных животных и животных с нокаутом генов, генетических заболеваний человека). Также это могут быть данные, применимые ко всей группе таких лекарственных препаратов (в том числе, подробная информация о биологических функциях молекулы-мишени и механизме действия, результаты исследований in vitro, исследований хронической токсичности или клинических исследований). В некоторых случаях имеющейся информации может быть достаточно для выяснения канцерогенного потенциала и установления риска для клинического применения без проведения дополнительных доклинических исследований.
Механизм действия некоторых биологических лекарственных препаратов может свидетельствовать о том, что они могут обладать канцерогенным потенциалом (например, иммунодепрессанты и факторы роста). Если совокупность различных данных (которые приведены выше) свидетельствует о существовании риска проявления канцерогенного потенциала, проведение биологических испытаний на грызунах не требуется. В такой ситуации наиболее приемлемым является указание о наличии риска в общей характеристике (инструкции по медицинскому применению (листке-вкладыше) данного лекарственного препарата, а также проведение мероприятий, направленных на минимизацию данного риска. В то же время если совокупность разных данных не позволяет сделать однозначный вывод, заявитель должен рассмотреть вопрос о проведении дополнительных исследований, которые могли бы оценить наличие риска, предполагаемого на основании знания механизма действия препарата (в соответствии с требованиями подраздела 4.8 главы 5.3 настоящих Правил).
Для ряда лекарственных препаратов сведений о свойствах и механизме их действия недостаточно для того, чтобы выдвинуть предположения о канцерогенном потенциале этих лекарственных препаратов. В таких случаях оправданной считается более тщательная оценка (например, оценка взаимосвязи между биологическими функциями молекулы-мишени и канцерогенным потенциалом или включение дополнительных конечных точек в токсикологические исследования препарата).
Если совокупность сведений, полученная в результате таких более обширных исследований, не указывает на наличие канцерогенного потенциала, проведение дополнительных доклинических исследований не рекомендуется. Напротив, если собранная информация свидетельствует о возможности проявления канцерогенного потенциала, заявитель может провести дополнительные доклинические исследования, которые позволили бы подтвердить отсутствие канцерогенного потенциала, в противном случае необходимо указать соответствующие предостережения в общей характеристике (инструкции по медицинскому применению (листке-вкладыше) данного лекарственного препарата.
Оценка канцерогенного потенциала, специфичная для данного препарата, используется для:
сообщения о наличии риска и составления плана по управлению рисками;
внесения соответствующих предостережений в общую характеристику (инструкцию по медицинскому применению (листок-вкладыш) данного лекарственного препарата;
обеспечения клинического мониторинга и пострегистрационного наблюдения.
Также могут использоваться сочетания вышеперечисленных подходов к обеспечению безопасности применения лекарственного препарата.
Информативность количественных исследований биологической активности на грызунах (или краткосрочных исследований канцерогенности) с использованием гомологичных белков при оценке канцерогенного потенциала препарата, который предполагается использовать в клинической практике, обычно ограничена.
Разработчику лекарственного препарата следует применять альтернативные решения по мере разработки новых подходов (методов) исследований безопасности лекарственных препаратов.
Примечания
1. Исследования перекрестной тканевой реактивности (ПТР (TCR)) представляют собой изучение связывания исследуемого препарата с тканями в тестах in vitro с использованием иммуногистохимических методик (ИГХ). Данные тесты позволяют охарактеризовать связывание препаратов моноклональных антител и препаратов модифицированных антител с антигенными детерминантами в тканях. Вместо иммуногистохимических методов могут быть использованы другие аналитические технологии, которые позволяют изучить распределение мишеней (участков связывания) в тканях.
Исследование ПТР с использованием панели тканей человека является рекомендуемым компонентом программы оценки безопасности, которая обосновывает применение начальной клинической дозы биотехнологического лекарственного препарата. Однако в отдельных случаях лекарственный препарат (кандидат для клинических исследований) не является хорошим реагентом, пригодным для использования в иммуногистохимических анализах, в связи с этим исследование ПТР может быть технически неосуществимо.
В исследованиях ПТР могут быть получены полезные дополнительные сведения о распределении мишени, а также может быть получена информация о возможном непредвиденном связывании. Само по себе связывание препарата с тканями не является показателем возможного проявления его биологической активности in vivo. Кроме того, связывание исследуемого препарата с участками, которые обычно являются недоступными для него в условиях in vivo (то есть с цитоплазмой), как правило, не имеет клинического значения. Именно поэтому результаты исследований следует интерпретировать, учитывая всю полученную информацию, включая фармакологические данные и данные по оценке безопасности препарата.
В случае непредвиденного связывания с тканями человека оценка ПТР выбранных тканей животных может обеспечить дополнительную информацию о потенциальных корреляциях либо их отсутствии в рамках доклинического исследования токсичности препарата. Исследования ПТР с использованием полного набора тканей животных не рекомендованы.
Поскольку лекарственные препараты на основе антител с двойной специфичностью (биспецифичные гибридные антитела) подлежат исследованию ПТР с использованием панели тканей человека, отпадает необходимость в изучении ПТР отдельных компонентов препарата.
Оценка связывания гомологичных белков с тканями является малоинформативной, если исследования ПТР препарата, предназначенного для клинических исследований, были проведены с использованием набора тканей человека, поэтому такая оценка не рекомендована.
Исследования ПТР не предназначены для выявления незначительных изменений критических показателей качества лекарственного препарата. Поэтому исследования ПТР не рекомендованы для оценки сопоставимости исследуемого препарата после внесения изменений в процесс его производства в ходе программы разработки препарата.
2. Если для оценки безопасности конъюгатов антител с лекарственными препаратами или токсинами (ADC) использовались 2 вида животных, необходимо провести дополнительное краткосрочное исследование (или часть краткосрочного исследования) с использованием неконъюгированного токсина по крайней мере на одном виде животных. В этих случаях предпочтительно использовать грызунов, за исключением случаев, когда токсин неактивен при введении грызунам. Если доступен только один фармакологически релевантный вид животных, исследования ADC следует проводить на данном виде животных. Выбор видов животных при исследовании новых токсических веществ, должен проводиться аналогично подходу, используемому при изучении новых химических веществ и основываться на индивидуальном подходе. В случае изучения токсинов или токсических веществ, которые не являются новыми и для которых доступно достаточное количество научной информации, проводить отдельное исследование неконъюгированного токсина не требуется. Должны быть представлены данные о сравнении метаболической стабильности ADC в организме животных и человека.
3. При интерпретации результатов исследования необходимо учитывать профиль воздействия препарата на развитие эмбриона и плода во время беременности. Белки с высокой молекулярной массой (> 5000 Да) не проникают через плацентарный барьер за счет простой диффузии. В случае моноклональных антител с высокой молекулярной массой (достигающей 150 000 Да) существует специфический транспортный механизм переносящий антитела через плаценту с участием неонатального Fc рецептора (FcRn), который определяет экспозицию у плода и отличается у разных биологических видов.
У НЧП и человека трансплацентарная передача IgG в период органогенеза невысока, она начинает увеличиваться в начале 2 триместра беременности, достигая максимума в конце 3 триместра. Проведение стандартных исследований эмбриофетальной токсичности на НЧП, получающих препарат с ранних сроков беременности и до 50-го дня гестации, не имеет большого значения при оценке непосредственного влияния на развитие эмбриона и плода в период органогенеза. В то же время такие исследования могут позволить оценить непрямое влияние на эмбриофетальное развитие в результате экспозиции препарата в организме матери. Кроме того, введение исследуемого препарата самкам НЧП после родов обычно неактуально, так как IgG экскретируются в грудное молоко только в раннем периоде (то есть в молозиво), в более поздних фазах лактации и период кормления грудным молоком его выделение прекращается.
Грызуны отличаются от НЧП и человека тем, что у них IgG проникают через желточный мешок благодаря транспорту с участием FcRn, в связи с этим экспозиция может возникать на относительно ранних сроках беременности по сравнению с НЧП и человеком. Кроме того, рождение потомства у грызунов происходят на этапе развития, когда они еще не достигли той степени зрелости, как у новорожденных НЧП и человека. Следовательно, чтобы детеныши экспериментальных животных подвергались экспозиции лекарственного препарата через молоко, самкам крыс (мышей) следует вводить препарат в период лактации по крайней мере до 9-го дня грудного вскармливания, когда зрелость потомства достигает того же уровня развития, что у новорожденного ребенка.
4. С целью проведения ранних функциональных исследований (например, оценка роста и поведения) минимальная продолжительность постнатального периода наблюдения за новорожденными после прекращения экспозиции препарата должна составлять 1 месяц.
В целом если существуют доказательства нежелательного влияния на иммунную систему или ее функции, полученные в ходе исследования общей токсичности, обосновано проведение исследований функций иммунной системы у потомства в послеродовый период в рамках исследований усиленной ППНР. При необходимости в раннем постнатальном периоде (до 28-го дня после рождения) следует провести иммунофенотипирование. Продолжительность постнатального наблюдения, целью которого является оценка функционального состояния иммунной системы, должна составлять 3 - 6 месяцев в зависимости от используемых функциональных тестов.
Нейроповеденческая оценка может ограничиваться наблюдениями за поведением в клинических условиях. Поскольку при обучении пользоваться предметами формирование навыков требует определенного времени, это может привести к продлению периода постнатального наблюдения по крайней мере до 9 месяцев и поэтому такой способ проверки не рекомендован.
5. Подробное обсуждение подхода к определению размеров группы макаков-крабоедов в исследовании усиленной ППНР представлено научной медицинской литературе. При проведении исследований усиленной ППНР число молодых животных должно быть достаточным (6 - 8 особей в группе в возрасте до 7 дней) для оценки постнатального развития и обеспечения возможности проведения специальных исследований (например, оценки состояния иммунной системы).
При проведении исследований усиленной ППНР, беременных животных набирают в течение нескольких недель или месяцев. Необходимо рассмотреть возможность прекращения дальнейшего набора в исследование беременных животных и коррекции дизайна исследования (например, путем использования кесарева сечения) в случаях, когда потери в пренатальном периоде в группе, получающей исследуемый препарат, свидетельствуют о токсических эффектах, связанных с воздействием препарата.
Рекомендуется повторно использовать самок животных контрольной группы, получавших индифферентное вещество, например, растворитель.
Если существуют основания предполагать, что за счет механизма действия препарат может оказывать влияние на эмбриофетальное развитие или приводить к прерыванию беременности, для подтверждения предполагаемой угрозы следует проводить исследования на ограниченном количестве животных.
6. В промежуточный отчет о результатах исследований, проведенных в рамках усиленной ППНР на НЧП, включают следующие конечные точки:
данные о самках (выживаемость, клинические наблюдения, масса тела, данные о воздействии препарата в период беременности (при наличии), любые фармакодинамические конечные точки);
данные о беременности (количество беременных животных, включенных в исследование, состояние беременности на момент завершения органогенеза (50-й день беременности) и на 100-й день беременности, частота невынашивания беременности и срок прерывания беременности). Для подготовки промежуточного отчета нет необходимости в проведении ультразвукового исследования с целью определения размера плода в связи с наличием сведений о фактической массе тела потомства при рождении;
данные об исходе беременности (число живо- и мертворожденных, масса тела новорожденных, выживаемость новорожденных и их масса тела на 7-е сутки после рождения, качественная оценка внешних морфологических признаков (подтверждение, что внешний вид соответствует пределам нормы), данные об экспозиции у потомства (при наличии), фармакодинамические конечные точки у потомства (если применимо)).
Глава 6. Спецификации. Методы испытания и критерии
приемлемости биотехнологических (биологических) препаратов
1. Введение
1.1. Цель
В настоящей главе представлены общие принципы формирования и обоснования единого свода спецификаций на биотехнологические и биологические препараты, представляемых в составе регистрационного досье для регистрации лекарственных препаратов.
1.2. Вводная часть
Под спецификацией понимается перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, представляющие собой численные (количественные) пределы, диапазоны и прочие критерии для описанных испытаний. Спецификация задает набор критериев, которым должны соответствовать активная фармацевтическая субстанция, лекарственный препарат или материалы других этапов производства, чтобы считаться приемлемыми для использования по целевому назначению. Спецификации составляются на все исходные материалы, сырье, промежуточные продукты, вспомогательные вещества, активную фармацевтическую субстанцию, лекарственный препарат. Для лекарственного препарата спецификация является частью нормативного документа по качеству. Под "соответствием спецификациям" понимается, что при испытании согласно представленным в спецификации аналитическим методикам активная фармацевтическая субстанция и лекарственный препарат будут отвечать заданным в спецификациях критериям приемлемости. Спецификации являются критическими стандартами качества, которые составляются и обосновываются производителем, после чего утверждаются уполномоченными органами государств-членов в качестве условий регистрации.
Спецификации являются частью общей стратегии контроля, разработанной для обеспечения качества лекарственных препаратов и постоянства характеристик лекарственных препаратов. Другие элементы данной стратегии включают:
подробное установление в процессе разработки характеристик (описание свойств), на основании которых составляются спецификации;
соответствие правилам надлежащей производственной практики Союза, утверждаемых Комиссией;
валидацию процесса производства;
испытание сырья и внутрипроизводственные испытания;
изучение стабильности и т.д.
Спецификации предназначены в большей мере для подтверждения качества активной фармацевтической субстанции и лекарственного препарата, чем для составления их полной характеристики с фармацевтической точки зрения, и должны описывать молекулярные и биологические свойства, контроль которых может быть использован для подтверждения безопасности и эффективности лекарственного препарата.
1.3. Сфера применения
Требования, изложенные в настоящей главе, применимы к лекарственным препаратам, которые являются белками и полипептидами, их производными, а также к лекарственным препаратам, компонентами которых они являются (например, конъюгатам). Такие белки и полипептиды продуцируются экспрессирующими системами рекомбинантных и нерекомбинантных клеточных культур и могут быть в надлежащей степени очищены и охарактеризованы с использованием подходящего набора аналитических методик.
Требования настоящей главы, можно также применять к другим лекарственным препаратам, например, белкам и полипептидам, изолированным из тканей и жидкостей организма. Для определения применимости настоящей главы в отношении таких лекарственных препаратов производители должны проконсультироваться с уполномоченными органами государств-членов. Если в других главах настоящих Правил указана ссылка на настоящую главу, то на такие лекарственные препараты распространяются требования, приведенные в настоящей главе, либо эти требования следует учитывать.
Требования настоящей главы не распространяются на антибиотики, синтетические пептиды и полипептиды, гепарины, витамины, клеточные метаболиты, препараты ДНК, экстракты аллергенов, традиционные вакцины, клетки, цельную кровь и клеточные компоненты крови.
В настоящей главе не приводятся требования к отдельным аналитическим методикам и частным критериям приемлемости. Кроме того, требования настоящей главы не предназначены для применения к спецификациям продуктов, проходящих стадии доклинического и (или) клинического исследования, однако приведенные в ней указания следует принимать во внимание для этих продуктов.
2. Принципы составления спецификаций
2.1. Установление характеристик (описание свойств)
Для составления надлежащих спецификаций необходимо с помощью надлежащих методов охарактеризовать свойства биологического или биотехнологического средства, включающие определение физико-химических свойств, биологической активности, иммунохимических свойств, чистоты и примесей. Критерии приемлемости следует устанавливать и обосновывать результатами анализа серий, использованных в доклинических и (или) клинических исследованиях, результатами анализа серий, использованных для подтверждения постоянства производства, результатами исследований стабильности и соответствующих данных по разработке.
Подробное установление характеристик выполняется на стадии разработки и, при необходимости, после внесения в процесс производства значительных изменений. На момент подачи документов на регистрацию препарат необходимо сравнить с соответствующим стандартным образцом (при наличии). По возможности (и если это применимо) препарат следует сравнить с его естественным аналогом. Кроме того, на момент подачи заявления о регистрации производитель должен располагать должным образом охарактеризованными собственными стандартными материалами, которые будут использоваться в ходе биологических и физико-химических испытаний промышленных серий. Разработчику лекарственного препарата следует учитывать, что постоянно разрабатываются новые аналитические технологии и модификации уже существующих технологий, которые необходимо применять при возникновении необходимости.
2.1.1. Физико-химические свойства.
Программа установления физико-химических свойств обычно включает определение состава, физических свойств и первичной структуры целевого (требуемого) продукта. В некоторых случаях с помощью соответствующих физико-химических методов можно получить данные о структурах более высокого порядка, правильность которых обычно подтверждается наличием биологической активности.
Поскольку продукция белков живыми организмами осуществляется с помощью процессов биосинтеза, таким белкам присуща определенная степень структурной гетерогенности, поэтому желаемый продукт может представлять собой смесь ожидаемых посттрансляционно модифицированных форм (например, гликоформ). Такие формы могут обладать активностью и не оказывать негативного влияния на безопасность и эффективность препарата (как это указано в подразделе 2.1.4 настоящей главы). Производитель должен установить профиль гетерогенности желаемого продукта и подтвердить его постоянство, путем испытания серий, использованных в доклинических и клинических исследованиях. Если установлен постоянный профиль гетерогенности продукта, оценка активности, эффективности и безопасности (включая иммуногенность) отдельных форм может не потребоваться.
Гетерогенность может также быть обусловлена производственными причинами и (или) хранением фармацевтической субстанции или лекарственного препарата. Поскольку гетерогенность таких препаратов определяет их качество, для обеспечения постоянства серий необходимо охарактеризовать степень и профиль такой гетерогенности. Если такие варианты целевого продукта обладают свойствами, сопоставимыми со свойствами самого целевого продукта по активности, эффективности и безопасности, эти варианты целевого продукта рассматривают в качестве родственных соединений. Если изменение процесса производства или продукты деградации приводят к появлению у продукта профилей гетерогенности, отличающихся от профиля гетерогенности для материала, использованного в доклинической и клинической разработке, необходимо изучить значимость таких изменений.
Аналитические методы, направленные на изучение физико-химических свойств, перечислены в подразделе 6.1 настоящей главы. Разработчику лекарственного препарата следует учитывать, что постоянно разрабатываются новые аналитические технологии и модификации уже существующих технологий, которые следует использовать, если это обосновано.
В целях выпускающего контроля качества серий (как это указано в разделе 4 настоящей главы) необходимо отобрать надлежащий перечень этих методов аналитического исследования и обосновать его.
2.1.2. Биологическая активность.
Оценка биологических свойств является не менее важным компонентом установления полного профиля свойств лекарственного препарата. Важным свойством лекарственного препарата является биологическая активность, которая определяет особую способность или свойство препарата оказывать определенный биологический эффект.
Производитель должен представить валидную биологическую методику количественного определения, позволяющую измерить биологическую активность. Примеры методик, используемых для определения биологической активности:
биологические методики количественного определения с использованием животных, которые определяют биологический ответ организма на препарат;
биологические методики количественного определения с использованием культур клеток, которые определяют биохимический или физиологический ответ на клеточном уровне;
биохимические методики количественного определения, которые определяют такие биологические активности, как скорость ферментативных реакций или биологические эффекты, обусловленные иммунологическими взаимодействиями.
Могут быть приемлемы и другие методики, такие как исследования лиганд-рецепторных взаимодействий.
Под активностью (potency) (выражаемой в единицах) понимается количественная мера биологической активности, основанная на показателе качества препарата, связанном с его значимыми биологическими свойствами, тогда как под количественным содержанием (quantity) (выражаемым в единицах массы) понимается физико-химическая мера содержания белка.
Не во всех случаях требуется воспроизводить биологическую активность в клинической ситуации. В ходе фармакодинамических и клинических исследований необходимо установить корреляцию между ожидаемым клиническим эффектом и активностью, рассчитанной с помощью биологической методики количественного определения.
Результаты количественного определения биологических методик следует выражать в единицах активности, откалиброванных по международному или национальному стандартному образцу (при их наличии и пригодности для использованной методики). Если такой стандартный образец отсутствует, необходимо приготовить собственный стандартный материал, а результаты испытания промышленных серий - выражать в разработанных единицах.
Физико-химические данные о сложных молекулах достаточно многообразны, тем не менее зачастую они не позволяют подтвердить структуру более высокого порядка, однако о ней можно косвенно судить по биологической активности. В таких случаях приемлема комбинация биологической методики с расширенными доверительными пределами и специальной количественной мерой. Следует отметить, что замена биологической методики количественного определения, измеряющей биологическую активность препарата, на физико-химические испытания допускается лишь при соблюдении 2 следующих условий:
с помощью таких физико-химических методов можно получить достаточно подробные физико-химические данные о продукте, включая сведения о структуре высокого порядка, при этом подтверждена соответствующая корреляция с биологической активностью;
у производителя лекарственного препарата имеется хорошо документированная история производства.
Если для расчета биологической активности, основанного на надлежащей корреляции, используются исключительно физико-химические испытания, их результаты следует выражать в единицах массы.
В целях выпускающего контроля качества серий (как это указано в подразделе 4 настоящей главы) производитель должен обосновать выбор соответствующей методики количественного определения (биологической и (или) физико-химической).
2.1.3. Иммунохимические свойства.
Если целевым продуктом является антитело, необходимо всесторонне описать его иммунологические свойства. Для определения аффинности, авидности и иммунореактивности (включая перекрестную реактивность) следует (по возможности) использовать методики связывания антитела с очищенными антигенами и определенными участками антигенов. Кроме того, необходимо описать биохимические свойства молекулы-мишени, несущей соответствующий эпитоп, а также сам эпитоп (по возможности).
В некоторых случаях молекулу белка некоторых активных фармацевтических субстанций и лекарственных препаратов необходимо изучить с помощью иммунохимических методик (например, ИФА, вестерн-блот), в которых используются антитела, распознающие различные эпитопы белковой молекулы. Иммунохимические свойства белка могут использоваться для установления его подлинности, постоянства свойств и чистоты или для количественного определения показателей качества.
Если оценка иммунохимических свойств используется в целях выпускающего контроля качества серий, необходимо представить все значимые сведения об антителе.
2.1.4. Чистота, примеси и контаминанты.
2.1.4.1. Чистота.
Установление абсолютной, равно как и относительной чистоты сопряжено со значительными аналитическими затруднениями, а результаты установления чистоты в значительной степени зависят от использованного метода. Традиционно относительную чистоту биологического препарата выражают с помощью специфической активности (в единицах биологической активности на миллиграмм препарата), которая также сильно зависит от использованного метода. Вследствие этого чистоту активной фармацевтической субстанции и лекарственного препарата изучают с помощью комбинации аналитических методик.
Ввиду уникальности биосинтетического процесса производства и молекулярных свойств биотехнологических и биологических препаратов активная фармацевтическая субстанция может состоять из нескольких молекулярных сущностей или вариантов. Если такие молекулярные сущности образуются в результате ожидаемых посттрансляционных модификаций, они являются частью целевого продукта. Если варианты целевого продукта образуются в ходе процесса производства и (или) хранения и обладают свойствами, сопоставимыми с целевым продуктом, их рассматривают в качестве родственных соединений, а не примесей (в соответствии с требованиями подраздела 2.1.1 настоящей главы).
Следует установить отдельные критерии приемлемости для отдельных родственных соединений или критерии приемлемости для их суммы.
В целях выпускающего контроля качества серий (в соответствии с требованиями подраздела 4 настоящей главы) для определения чистоты необходимо отобрать и обосновать подходящий перечень методов.
2.1.4.2. Примеси.
Помимо изучения чистоты активной фармацевтической субстанции и лекарственного препарата, которые могут состоять из целевого продукта и множества родственных соединений, производитель также должен изучить примеси, которые могут в них содержаться. Примеси делятся на производственные и родственные. Они могут иметь изученную структуру, могут быть описанными частично или неидентифицированными. Если удается получить достаточное количество примесей, их необходимо как можно подробнее охарактеризовать и по возможности изучить их биологическую активность.
Производственные примеси образуются в ходе процесса производства, например, из клеточных субстратов (например, белки клетки-хозяина, ДНК клетки-хозяина), клеточной культуры (например, индукторы, антибиотики или компоненты питательной среды) или вследствие последующей переработки (в соответствии с требованиями подраздела 6.2.1 настоящей главы). Родственные примеси (например, прекурсоры, определенные продукты деградации) - это молекулярные варианты, образующиеся при производстве и хранении целевого продукта, которые не обладают активностью, эффективностью и безопасностью сопоставимыми с целевым продуктом.
Кроме того, критерии приемлемости по содержанию примесей в целевом продукте должны основываться на результатах анализа серий, использованных в доклинических и клинических исследованиях, и серий, испытанных для подтверждения постоянства производства.
Следует установить отдельные критерии приемлемости для отдельных родственных примесей или критерии приемлемости для их суммы. В определенных случаях для некоторых примесей критерии приемлемости устанавливать не требуется (в соответствии с требованиями подраздела 2.3 настоящей главы).
Примерный перечень аналитических методик, которые целесообразно использовать в испытаниях на примеси, приведен в подразделе 6.2 настоящей главы. Разработчику лекарственного препарата следует учитывать, что непрерывно разрабатываются новые аналитические технологии и модификации ныне существующих аналитических технологий. При наличии достаточного обосновании их также следует использовать для контроля качества целевого продукта.
В целях выпускающего контроля качества серий (в соответствии с требованиями подраздела 4 настоящей главы) необходимо отобрать надлежащий перечень методов контроля качества и обосновать его.
2.1.4.3. Контаминанты
К контаминантам препарата относятся все посторонним образом привнесенные материалы, не используемые в процессе производства, например, химические и биохимические материалы (например, микробные протеазы) и (или) микроорганизмы. Необходимо избегать наличия контаминантов и (или) надлежащим образом контролировать их содержание с помощью надлежащих внутрипроизводственных критериев приемлемости или пределов действия в спецификациях на фармацевтическую субстанцию или лекарственный препарат (в соответствии с требованиями подраздела 2.3 настоящей главы). При особых случаях контаминации посторонними вирусами или микоплазмой концепция пределов действия неприменима, поэтому необходимо следовать стратегиям, указанным в главах 1 и 2 настоящих Правил.
2.1.5. Количественное содержание.
Количественная характеристика препарата, определяемая содержанием белка, имеет большое значение для биотехнологических и биологических препаратов и определяется с помощью соответствующих методов анализа (обычно физико-химических). В частных случаях допускается подтвердить, что установленные такими методами показатели качества могут быть непосредственно связаны с показателями, определенными в ходе биологического анализа. При наличии такой корреляции целесообразнее измерять количество, а не биологическую активность в ходе таких производственных процессов, как наполнение.
2.2. Аналитические вопросы
2.2.1. Стандартные образцы и стандартные материалы
При регистрации новых молекулярных соединений международные или национальные стандарты, как правило, будут отсутствовать. На момент подачи заявления о регистрации лекарственного препарата производитель должен разработать надлежащим образом охарактеризованный собственный основный стандартный материал, приготовленный из серий, отражающих свойства промышленного и клинического материалов. Собственные рабочие стандартные материалы, используемые для испытания промышленных серий, следует калибровать по основному стандартному материалу. При наличии международного или национального стандарта и его пригодности собственные стандартные материалы следует калибровать по нему. Хотя как в биологических методиках, так и физико-химических испытаниях рекомендуется использовать одинаковый стандартный материал, в некоторых случаях необходимо использовать различные стандартные материалы. Кроме того, могут потребоваться отдельные стандартные материалы для родственных соединений, родственные и производственные примеси. В соответствующих случаях в регистрационное досье необходимо включить описание производства и (или) очистки стандартных материалов. Необходимо также представить документацию по характеристикам, условиям хранения и составу стандартных материалов, обосновывающую стабильность стандартных материалов.
2.2.2. Валидация аналитических методик
При подаче заявления о регистрации лекарственного препарата в уполномоченные органы государств-членов заявители должны валидировать аналитические методики, использованные в спецификациях, в соответствии с требованиями актов, входящих в право Союза, за исключением отдельных методик, присущих исключительно испытаниям, используемым для анализа биотехнологических и биологических лекарственных препаратов.
2.3. Контроль производства
2.3.1. Особенности технологического процесса.
Правильное планирование процесса производства и теоретический анализ его возможностей являются частью стратегии разработки контролируемого и воспроизводимого процесса производства, позволяющего получить активную фармацевтическую субстанцию и лекарственный препарат, отвечающие требованиям спецификации. В этом отношении пределы отклонений показателей качества обосновывают критическими данными, полученными на протяжении всего процесса производства (начиная с раннего этапа разработки и заканчивая промышленным производством).
Испытания активной фармацевтической субстанции или лекарственного препарата на определенные примеси могут не потребоваться (их допускается не включать в спецификации), если с помощью подходящих исследований подтверждены эффективный контроль их содержания или приемлемая степень очистки. Такие испытания могут включать верификацию промышленных серий в соответствии с требованиями, содержащимися в актах, входящих в право Союза. На момент регистрации в распоряжении заявителя могут находиться лишь ограниченные данные по верификации промышленных серий, поэтому данную концепцию в частных случаях допускается внедрять на пострегистрационном этапе в соответствии с требованиями, содержащимися в актах, входящих в право Союза.
2.3.2. Внутрипроизводственные критерии приемлемости и уровень действия.
Внутрипроизводственные испытания проводятся на этапах принятия критических решений, а также на других этапах, если полученные результаты испытаний указывают на необходимость удостовериться в постоянстве процесса производства активной фармацевтической субстанции или лекарственного препарата. Результаты внутрипроизводственных испытаний регистрируют в виде пределов уровня действия или критериев приемлемости. Проведение таких испытаний позволяет исключить испытания фармацевтической субстанции или лекарственного препарата (в соответствии с требованиями подраздела 2.3.1 настоящей главы). Внутрипроизводственные испытания in vitro на посторонние агенты клеток предельного для производства клеточного возраста являются примером испытания, для которого необходимо установить критерии приемлемости.
Производитель также должен использовать собственные (внутренние) пределы уровня действия для оценки процесса на менее значимых этапах. Основой для разработки предварительных пределов уровня действия, устанавливаемых для процесса производства, должны служить данные, полученные в ходе разработки лекарственного препарата и проведения валидационных циклов. Такие пределы, ответственность за установление которых несет производитель, могут быть использованы для начала расследования возможных отклонений или дальнейших действий, направленных на их устранение. В дальнейшем эти пределы следует изучать и корректировать по мере приобретения дополнительного производственного опыта и данных после регистрации лекарственного препарата.
2.3.3. Спецификации на исходные материалы, сырье и вспомогательные вещества.
Качество исходных материалов и сырья, использовавшегося в производстве фармацевтической субстанции или лекарственного препарата, должно удовлетворять стандартам, соответствующим их целевому назначению. Исходные материалы и сырье (включая реактивы) биологического происхождения требуют тщательного изучения на предмет наличия или отсутствия опасных эндогенных и посторонних агентов. Методики, позволяющие использовать аффинную хроматографию (например, применение моноклональных антител), должны включать соответствующие меры, обеспечивающие отсутствие негативного влияния таких производственных примесей и потенциальных контаминантов, образующихся в ходе производства, на качество и безопасность фармацевтической субстанции и лекарственного препарата. Необходимо представить соответствующие сведения об использующихся антителах.
По возможности качество вспомогательных веществ, использующихся в производстве лекарственного препарата (и в некоторых случаях, фармацевтической субстанции), а также система "контейнер - укупорка" должны удовлетворять фармакопейным стандартам. Иначе, для нефармакопейных вспомогательных веществ, необходимо установить удовлетворительные критерии приемлемости.
2.4. Фармакопейные спецификации
Фармакопеи государств-членов и основные фармакопеи, в соответствии с Концепцией гармонизации фармакопей государств - членов Евразийского экономического союза, утвержденной Решением Коллегии Комиссии от 22 сентября 2015 г. N 119 (далее - фармакопеи) содержат в фармакопейных статьях (монографиях) важные требования в отношении определенных аналитических методик и критериев приемлемости, которые, если они применимы, являются частью процесса оценки фармацевтической субстанции либо лекарственного препарата. Такие фармакопейные статьи (монографии), применимые к биотехнологическим и биологическим препаратам, включают в том числе испытания на стерильность, эндотоксины, микробиологическую чистоту, объем продукта в контейнере, однородность дозирования и механические включения. С точки зрения использования фармакопейных методов и критериев приемлемости требования настоящей главы связаны с обеспечением определенной степени гармонизации аналитических методик между фармакопеями. Фармакопеи предназначены для разработки идентичных или методологически эквивалентных аналитических методик и критериев приемлемости.
2.5. Допустимые пределы на выпуск и допустимые пределы
на срок годности
При обосновании показателей качества допускается использовать концепцию допустимых пределов на выпуск вместо допустимых пределов на срок годности. Эта концепция заключается в установлении допустимых пределов показателей качества активной фармацевтической субстанции или лекарственного препарата, которые на момент их выпуска более строгие, чем на срок годности. Примерами применения концепции могут служить активность (potency) и продукты деградации. В частных случаях концепцию пределов на выпуск допускается применять исключительно в отношении собственных допустимых пределов, но не установленных в регистрационном досье допустимых пределов на срок годности.
2.6. Статистические концепции
При необходимости представления количественных данных следует проводить надлежащий статистический анализ. Необходимо всесторонне описать методы анализа, включая их обоснование и научные предпосылки выбора. Это описание должно быть достаточно полным (ясным), чтобы выполнить независимую обработку представленных результатов.
3. Обоснование спецификации
Составление спецификаций на фармацевтическую субстанцию и лекарственный препарат является частью общей стратегии контроля качества, включающей контроль качества исходных материалов, сырья и вспомогательных веществ, внутрипроизводственные испытания, оценку или валидацию процесса производства, соблюдение правил надлежащей производственной практики Союза, утверждаемых Комиссией, изучение стабильности и испытание постоянства свойств серий. В совокупности указанные элементы обеспечивают надлежащее качество лекарственного препарата. Поскольку спецификации предназначены в большей мере для подтверждения качества, а не для характеристики свойств лекарственного препарата и (или) активной фармацевтической субстанции, производитель должен представить предпосылки выбора и обоснование включения в спецификацию и (или) исключения из нее испытаний определенных показателей качества. При разработке научно обоснованных спецификаций необходимо учитывать ряд аспектов, указания по которым представлены ниже.
Спецификации должны быть связаны с процессом производства. Спецификации должны основываться на результатах анализа серий, использованных для подтверждения постоянства производства. Необходимо увязать спецификации и процесс производства, особенно по родственным соединениям, родственным и производственным примесям. Изменения процесса производства и продукты деградации, образующиеся при хранении, могут приводить к гетерогенным профилям, отличающимся от профилей у материала, использованного в доклинической и клинической разработке. Необходимо изучить значимость таких изменений.
Спецификации должны позволять провести оценку стабильности активной фармацевтической субстанции и лекарственного препарата. При разработке спецификаций необходимо учитывать деградацию (старение) активной фармацевтической субстанции и лекарственного препарата, которая происходит при хранении. Ввиду присущей этим продуктам сложной структуры невозможно подобрать одну методику или один параметр, описывающие профиль стабильности. Вследствие этого производитель должен предложить в регистрационном досье профиль показателей стабильности. Результаты испытания такого профиля показателей стабильности должны в дальнейшем позволять обнаружить изменения качества лекарственного препарата и (или) активной фармацевтической субстанции. Перечень включаемых в спецификацию испытаний зависит от свойств лекарственного препарата. Производителю следует выполнять требования главы 8 настоящих Правил.
Спецификации должны быть связаны с доклиническими и клиническими исследованиями. Спецификации необходимо составлять, основываясь на результатах анализа серий, использованных в доклинических и клинических исследованиях. Качество материала, производимого в промышленном масштабе, должно соответствовать качеству серий, использованных в доклинических и клинических исследованиях.
Спецификации должны быть связаны с аналитическими методиками. Критические показатели качества включают, например, биологическую активность (potency), свойства и количество родственных соединений, родственных и производственных примесей. Указанные показатели изучают с помощью большого числа аналитических методик, каждая из которых характеризует различные свойства препарата. При разработке препарата нередко вместе с совершенствованием технологии производства препарата происходит совершенствование аналитической технологии. В связи с этим необходимо подтвердить, что данные, полученные в ходе разработки, коррелируют с данными, полученными на момент подачи заявления о регистрации лекарственного препарата.
4. Спецификации
Выбор испытаний, включаемых в спецификации, зависит от лекарственного препарата. Необходимо описать научные предпосылки, на основании которых был установлен допустимый диапазон критериев приемлемости. Критерии приемлемости следует устанавливать и обосновывать, руководствуясь результатами анализа серий, использованных в доклинических и (или) клинических исследованиях, результатами анализа серий, использованных для подтверждения постоянства производства, результатов исследований стабильности и релевантных данных по разработке лекарственного препарата.
В некоторых случаях допускается проводить испытания на промежуточных продуктах, а не фармацевтической субстанции или лекарственном препарате. В таких случаях результаты испытаний следует рассматривать в качестве внутрипроизводственных критериев приемлемости и включать их в спецификацию на фармацевтическую субстанцию или лекарственный препарат, если это предусмотрено требованиями уполномоченных органов государств-членов.
4.1. Спецификация на активную фармацевтическую субстанцию
Приведенные в настоящем подразделе главы испытания и критерии приемлемости, в целом, применимы ко всем активным фармацевтическим субстанциям (соответствующие аналитические методики указаны в подразделе 2.2.2 настоящей главы). В применимых случаях с активной фармацевтической субстанцией необходимо проводить фармакопейные испытания (например, определение эндотоксинов). В частных случаях могут потребоваться также дополнительные критерии приемлемости, специфичные для активной фармацевтической субстанции.
4.1.1. Внешний вид и описание.
Необходимо дать качественное описание физического состояния (например, твердое, жидкое) и цвета активной фармацевтической субстанции.
4.1.2. Подлинность.
Испытания на подлинность должны быть высоко специфичными в отношении фармацевтической субстанции и основываться на уникальных свойствах ее молекулярной структуры и (или) иных особенностях. Для установления подлинности может потребоваться проведение более одного испытания (физико-химического, биологического и (или) иммунохимического). Испытания могут носить качественный характер. В целях установления подлинности допускается использовать и (или) соответствующим образом модифицировать методы, традиционно используемые для установления свойств препарата и описанные в подразделах 2.1 и 6.1 настоящей главы.
4.1.3. Чистота и примеси.
Абсолютную чистоту биотехнологических и биологических препаратов сложно установить, а результаты ее определения зависят от использованного метода (в соответствии с указаниями подраздела 2.1.4 настоящей главы). Вследствие этого чистоту фармацевтической субстанции, как правило, оценивают с помощью комбинации методов. При выборе и оптимизации аналитических методик следует стремиться к отделению целевого продукта от родственных соединений и примесей.
Примеси таких препаратов делятся на производственные и родственные:
к производственным примесям (в соответствии с требованиями подраздела 2.1.4 настоящей главы) активной фармацевтической субстанции относятся питательные среды, белки и ДНК клетки-хозяина, моноклональные антитела, хроматографические среды, использованные при очистке, растворители и компоненты буферных растворов. Минимизировать содержание таких примесей необходимо путем выполнения надлежащего контроля производственных процессов;
родственные примеси (в соответствии с требованиями подраздела 2.1.4 настоящей главы) активной фармацевтической субстанции - это молекулярные варианты, образующиеся при производстве и (или) хранении и отличающиеся от целевого продукта по своим свойствам.
При выборе и оптимизации аналитических методик испытания на примеси следует стремиться к отделению целевого продукта и родственных соединений от примесей. Необходимо разработать критерии приемлемости на отдельные примеси и (или) на сумму примесей. В частных случаях критерии приемлемости на некоторые примеси могут не потребоваться (в соответствии с требованиями подраздела 2.3 настоящей главы).
4.1.4. Активность (potency).
В спецификации необходимо включить подходящую валидированную методику испытания на активность (в соответствии с требованиями подраздела 2.1.2 настоящей главы) биотехнологических или биологических активных фармацевтической субстанции и (или) лекарственного препарата. Если для испытания на подлинность лекарственного препарата (в соответствии с требованиями подраздела 4.2.4 настоящей главы) используется надлежащая методика, то для количественного определения активной фармацевтической субстанции может быть достаточным альтернативный метод (физико-химический и (или) биологический). В некоторых случаях дополнительную ценность могут представлять результаты определения специфической активности.
4.1.5. Количественное содержание (quantity).
Необходимо определить количественное содержание фармацевтической субстанции, основанное, как правило, на содержании (массе) белка, используя надлежащую методику. Определение количественного содержания может осуществляться без стандартного образца или материала. Если производство лекарственного препарата основывается на биологической активности, дополнительное определение количественного содержания в частных случаях не требуется.
4.2. Спецификация на лекарственный препарат
Следующие испытания и критерии приемлемости в целом применимы ко всем лекарственным препаратам. Каждый подраздел 4.2.1 - 4.2.5 настоящей главы по лекарственному препарату содержит перекрестные ссылки на подразделы 4.1.1 - 4.1.5 настоящей главы по активной фармацевтической субстанции. К соответствующим лекарственным формам применяются фармакопейные требования Союза и государств-членов. К стандартным испытаниям, указанным в фармакопеях, относятся в том числе стерильность, эндотоксины, микробиологическая чистота, извлекаемый объем, механические включения, однородность единиц дозирования, содержание воды (влаги) в лиофилизате. Если применимо, испытание на однородность единиц дозирования допускается проводить в качестве внутрипроизводственного контроля, устанавливая при этом соответствующие критерии приемлемости.
4.2.1. Внешний вид и описание.
Необходимо привести качественное описание физического состояния (например, твердое, жидкое), цвета и прозрачности лекарственного препарата.
4.2.2. Подлинность.
Испытания на подлинность должны быть высоко специфичными в отношении лекарственного препарата и основываться на уникальных свойствах его молекулярной структуры и (или) иных особенностях. По своей природе испытания на подлинность могут быть качественными. Несмотря на то что в большинстве случаев достаточно одного испытания, для установления подлинности некоторых препаратов необходимо провести несколько испытаний (физико-химических, биологических и (или) иммунохимических). В целях установления подлинности лекарственного препарата допускается использовать и (или) соответствующим образом модифицировать методы, традиционно используемые для описания свойств активной фармацевтической субстанции и указанные в подразделах 2.1 и 6.1 настоящей главы.
4.2.3. Чистота и примеси.
При производстве и (или) хранении лекарственного препарата могут образовываться примеси или может увеличиваться их содержание. Примеси могут совпадать с примесями активной фармацевтической субстанции, то есть быть производственными, либо быть продуктами деградации, образующимися исключительно в лекарственном препарате при его приготовлении (формуляции) или хранении. Если качественное и количественное содержание примесей (то есть их относительные количества и (или) концентрации) совпадает с таковым у активной фармацевтической субстанции, включение в спецификацию дополнительных испытаний не требуются. Если известно, что примеси привносятся или образуются при производстве и (или) хранении лекарственного препарата, необходимо определить их содержание и установить критерии их приемлемости.
В целях определения изменений активной фармацевтической субстанции при производстве и (или) хранении лекарственного препарата критерии приемлемости и аналитические методики необходимо разрабатывать и обосновывать с учетом предыдущего опыта изучения лекарственного препарата.
При выборе и оптимизации аналитических методик определения примесей следует стремиться к отделению целевого продукта и родственных соединений от примесей, включая продукты деградации, и вспомогательных веществ.
4.2.4. Активность (potency).
В спецификации необходимо включить подходящую валидированную методику испытания на активность (как это указано в подразделе 2.1.2 настоящей главы) биотехнологических или биологических активной фармацевтической субстанции и (или) лекарственного препарата. Если для испытания на подлинность активной фармацевтической субстанции используется надлежащая методика, то для количественного определения лекарственного препарата может быть достаточным альтернативный метод (физико-химический и (или) биологический). Однако необходимо представить обоснование такого выбора.
4.2.5. Количественное содержание (quantity).
Необходимо определить количественное содержание активной фармацевтической субстанции в лекарственном препарате, основанное, как правило, на содержании (массе) белка, используя надлежащую методику. Если производство лекарственного препарата основывается на биологической активности, дополнительное определение количественного содержания в частных случаях может не потребоваться.
4.2.6. Общие испытания.
Зачастую для оценки функциональных свойств лекарственного препарата необходимо охарактеризовать его физические свойства и определить иные показатели качества лекарственного препарата. Примерами таких испытаний служат pH и осмолярность.
4.2.7. Дополнительное испытание уникальных лекарственных форм.
Выпуск лекарственного препарата в определенных лекарственных формах может потребовать проведения дополнительных, связанных с видом такой лекарственной формы испытаний, неупомянутых выше.
5. Определения
Для целей настоящей главы используются понятия (термины), которые означают следующее:
"активная фармацевтическая субстанция (нерасфасованный материал)" - материал, который впоследствии смешивается со вспомогательными веществами для получения лекарственного препарата. Он может состоять из целевого продукта, родственных соединений и родственных и производственных примесей. Он также может содержать вспомогательные вещества, включая иные компоненты, например, буферные растворы;
"активность" - мера биологической активности, определяемая с помощью подходящей биологической методики количественного определения (также называемой методикой количественного определения активности (potency assay) или биометодикой (bioassay)), основанная на показателе качества продукта, который связан с релевантными биологическими свойствами;
"биологическая активность" - особая способность или свойство препарата оказывать определенный биологический эффект. Количественной мерой биологической активности является активность (potency);
"вспомогательное вещество" - компонент, преднамеренно добавляемый к активной фармацевтической субстанции, который в прибавляемых количествах не должен обладать фармакологическими свойствами;
"контаминанты" - любые привнесенные посторонние материалы (например, химические, биохимические или микробные), не предусмотренные процессом производства активной фармацевтической субстанции или лекарственного препарата;
"критерии приемлемости" - численные пределы, диапазоны и другие подходящие меры приемлемости результатов аналитических методик, которым должны удовлетворять активная фармацевтическая субстанция, лекарственный препарат или материалы других этапов производства;
"лекарственный препарат (лекарственная форма, готовый препарат)" - разновидность фармацевтического продукта, который содержит активную фармацевтическую субстанцию, как правило, в комбинации со вспомогательными веществами;
"предел действий (предел, требующий принятия мер)" - собственное значение, используемое для оценки постоянства процесса производства на менее критических этапах;
"примесь" - любой компонент, содержащийся в активный фармацевтической субстанции или лекарственном препарате, не являющийся целевым продуктом, родственным соединением или вспомогательным веществом, включая буферные компоненты. Различают производственные и родственные примеси;
"продукты деградации" - молекулярные варианты, образующиеся вследствие изменений целевого продукта или родственных соединений со временем и (или) под воздействием, например, света, температуры, pH, влаги, или вследствие взаимодействия со вспомогательным веществом и (или) первичной системой "контейнер - укупорка". Такие изменения могут возникать при производстве и (или) хранении (например, дезаминирование, окисление, агрегация, протеолиз). Продукты деградации могут представлять собой родственные соединения либо родственные примеси;
"производственные примеси" - примеси, образующиеся в процессе производства. Они могут проистекать из клеточных субстратов (например, белки клетки-хозяина, ДНК клетки-хозяина), клеточной культуры (например, индукторы, антибиотики или компоненты питательной среды) или последующей обработки (например, реактивы для обработки или экстрагируемые (вымываемые) из колонки вещества);
"родственные примеси" - молекулярные варианты целевого продукта (например, прекурсоры, некоторые продукты деградации, образующиеся при производстве и (или) хранении), которые не обладают сопоставимыми с целевым продуктом активностью, эффективностью и безопасностью;
"родственные соединения" - молекулярные варианты целевого продукта, образующиеся при производстве и (или) хранении, обладающие активностью и не имеющие негативного влияния на безопасность и эффективность лекарственного препарата. Эти варианты обладают свойствами, сопоставимыми с целевым продуктом, и не рассматриваются в качестве примесей;
"собственный основной стандартный материал" - надлежащим образом охарактеризованный материал, приготовленный производителем из репрезентативных серий в целях проведения биологических методик или физико-химических испытаний последующих серий, по которому также калибруют собственный рабочий стандартный материал;
"собственный рабочий стандартный материал" - материал, приготовленный аналогично основному стандартному материалу и предназначенный исключительно для анализа и контроля отдельных показателей качества последующих серий. Во всех случаях его калибруют по собственному основному стандартному материалу;
"спецификация" - перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, представляющие собой численные (количественные) пределы, диапазоны и прочие критерии для описанных испытаний. Она задает набор критериев, которым должны соответствовать фармацевтическая субстанция, лекарственный препарат или материалы других этапов производства, чтобы считаться приемлемыми для использования по целевому назначению. "Соответствие спецификациям" - способность фармацевтической субстанции и лекарственного препарата, подвергшихся испытаниям согласно описанным в спецификациях аналитическим методикам, удовлетворять перечисленным в них критериям приемлемости. Спецификации являются ключевыми стандартами качества, которые предлагаются и обосновываются производителем и утверждаются уполномоченным органом в качестве условий регистрации;
"целевой продукт" - 1) белок, обладающий ожидаемой структурой; 2) белок, ожидаемый на основании последовательности ДНК и предполагаемой посттрансляционной модификации (включая гликоформы) и на основании предлагаемой последующей обработки (после культивирования), направленной на получение активной (действующей) биологической молекулы.
6. Дополнение
6.1. Характеристика физико-химических свойств
Настоящее дополнение содержит указания по техническим подходам, которые позволяют охарактеризовать и подтвердить структуру целевого продукта, активной фармацевтической субстанции и (или) лекарственного препарата и проанализировать их физико-химические свойства. Используемые подходы будут отличаться от препарата к препарату, во многих случаях потребуется придерживаться подходов, не включенных в настоящее дополнение. Непрерывно возникают новые аналитические технологии и модификации ныне существующих. При достаточном обосновании их также следует использовать.
6.1.1. Характеристика и подтверждение структуры.
Аминокислотная последовательность. Необходимо, насколько это возможно, установить аминокислотную последовательность целевого продукта, используя подходы, подобно описанным в абзацах втором - пятом настоящего подраздела, и затем сравнить их с аминокислотной последовательностью, которая соответствует нуклеотидной последовательности гена целевого продукта.
Аминокислотный состав. Общий аминокислотный состав определяют, используя различные гидролитические и аналитические методики, и сравнивают с аминокислотной последовательностью, которая соответствует нуклеотидной последовательности гена целевого продукта. Во многих случаях анализ аминокислотного состава позволяет получить определенные ценные сведения о структуре пептидов и небольших белков, однако для больших белков эти данные менее точны. Во многих случаях результаты количественного аминокислотного анализа позволяют установить содержание белка.
Терминальные последовательности аминокислот. Анализ концевых аминокислот проводят для установления природы и однородности амино- и карбси-концевых аминокислот. Если целевой продукт является гетерогенным по концевым аминокислотам, с помощью соответствующей аналитической методики необходимо определить относительное содержание вариантных форм. Последовательность таких концевых аминокислот необходимо сравнить с последовательностью концевых аминокислот, соответствующей нуклеотидной последовательности гена целевого продукта.
Пептидная карта. Избирательная фрагментация препарата на отдельные пептиды осуществляется при помощи подходящих ферментов или химических соединений, а образующиеся пептидные фрагменты подвергаются анализу с помощью ВЭЖХ или иной подходящей аналитической методики. Используя такие методы, как анализ аминокислотного состава, N-концевое секвенирование или масс-спектрометрию, необходимо, насколько это возможно, идентифицировать пептидные фрагменты. Пептидное картирование активной фармацевтической субстанции или лекарственного препарата с помощью должным образом валидированной методики - это метод, часто использующийся для подтверждения структуры целевого продукта в целях выпускающего контроля качества серий.
Сульфгидрильные группы и дисульфидные мостики. Если согласно данным нуклеотидной последовательности гена целевого продукта предполагается наличие остатков цистеина, необходимо по возможности определить количество и положения всех свободных сульфгидрильных групп и (или) дисульфидных мостиков. С этой целью используют пептидное картирование (в восстанавливающих и невосстанавливающих условиях), масс-спектрометрию или иные подходящие технологии.
Углеводная структура. Необходимо определить содержание углеводов в гликопротеинах (нейтральные сахара, аминосахара и сиаловые кислоты). Кроме того, необходимо по возможности проанализировать структуру углеводных цепей, олигосахаридный профиль (профиль ветвления) и участки гликозилирования полипептидной цепи.
6.1.2. Физико-химические свойства.
Молекулярная масса или размер. Молекулярную массу или размер определяют с помощью эксклюзионной хроматографии, электрофореза в полиакриламидном геле с натрия додецилсульфатом (в восстанавливающих и (или) невосстанавливающих условиях), масс-спектрометрии и прочих подходящих технологий.
Профиль изоформ. Определяют с помощью изоэлектрического фокусирования или иных подходящих технологий.
Коэффициент экстинкции или молярный коэффициент поглощения. Во многих случаях целесообразно определять коэффициент экстинкции или молярного поглощения целевого продукта при определенной длине волны в ультрафиолетовом (УФ) или видимом спектре (например, 280 нм). Коэффициент экстинкции определяют с помощью УФ-спектрофотометрии (спектрофотометрии) в видимом диапазоне раствора препарата, содержащего известное количество белка, рассчитанного с помощью таких методов, как анализ аминокислотного состава или определение содержания азота и т.д. Если для измерения содержания белка используется УФ-абсорбция, следует использовать коэффициент экстинкции этого препарата.
Электрофоретические профили. Электрофоретические профили, данные о подлинности, однородности и чистоте получают с помощью электрофореза в полиакриламидном геле, изоэлектрического фокусирования, электрофореза в полиакриламидном геле с натрия додецилсульфатом, вестерн-блота, капиллярного электрофореза и прочих подходящих методов.
Профили жидкостной хроматографии. Профили жидкостной хроматографии, данные о подлинности, однородности и чистоте получают с помощью эксклюзионной хроматографии, обращенно-фазной жидкостной хроматографии, ионо-обменной жидкостной хроматографии, аффинной хроматографии и прочих подходящих методов.
Спектроскопические профили. Определяют спектры абсорбции в ультрафиолетовом и видимом диапазонах. Структуры высокого порядка изучают с помощью таких методов, как круговой дихроизм, ядерный магнитный резонанс (ЯМР) или другие подходящие технологии.
6.2. Дополнение по примесям
В настоящем подразделе указаны потенциальные примеси, их источники и примеры применимых аналитических подходов их обнаружения. Подобно характеристике физико-химических свойств, профиль примесей и использующиеся технические подходы отличаются от препарата к препарату; во многих случаях необходимо придерживаться подходов, не включенных в настоящее приложение. Разработчику лекарственного препарата следует учитывать, что непрерывно возникают новые аналитические технологии и модификации ныне существующих. При достаточном обосновании их также следует использовать.
6.2.1. Производственные примеси и контаминанты.
Образуются в ходе процесса производства (пункт 2.1.4 настоящей главы) и делятся на 3 основные категории: проистекающие из клеточного субстрата, клеточной культуры и вследствие последующей обработки.
К проистекающим из клеточного субстрата примесям относятся белки организма-хозяина, нуклеиновая кислота (геномная, векторная или вся ДНК клетки-хозяина), но ими не ограничиваются. Чувствительными к белкам клетки-хозяина методиками являются, например, иммунологические методы, способные обнаруживать широкий диапазон белковых примесей. Поликлональные антитела, используемые в иммунологическом методе, получают путем иммунизации животных препаратом клеток-продуцентов, не содержащих ген, кодирующий целевой продукт, препаратом партнеров гибридизации (фузии) и препаратом иных подходящих клеточных линий. Содержание ДНК клеток хозяина можно определить с помощью прямого анализа препарата (например, с помощью технологий гибридизации). Во избежание установления критериев приемлемости для этих примесей в некоторых случаях подтверждают удаление примесей, проистекающих из клеточного субстрата (например, нуклеиновых кислот и белков клетки-хозяина), для этого прибегают к исследованиям клиренса, которые могут включать в себя лабораторные эксперименты с добавлением таких примесей.
К проистекающим из клеточной культуры примесям относятся индукторы, антибиотики, сыворотка и прочие компоненты питательных сред, но ими не ограничиваются.
К примесям, образующимся при последующей обработке, относятся ферменты, химические и биологические реактивы для обработки (например, бромциан, гуанидин, окисляющие и восстанавливающие вещества), неорганические соли (например, тяжелые металлы, мышьяк, неметаллические ионы), растворители, носители, лиганды (например, моноклональные антитела) и прочие экстрагируемые вещества, но ими не ограничиваются.
Необходимо в соответствии с главой 2 настоящих Правил подтвердить способность процесса производства элиминировать и (или) инактивировать намеренно привнесенные, эндогенные и посторонние вирусы.
6.2.2. Родственные примеси, включая продукты деградации.
Ниже представлены наиболее часто встречающиеся варианты целевого продукта и перечень соответствующих технологий их анализа. Указанные варианты могут потребовать значительных усилий для их выделения и характеристики их свойств с целью установления вида модификаций. Используя должным образом установленные критерии приемлемости, необходимо провести испытания и контролировать содержание продуктов деградации, в значительных количествах образующихся при производстве и (или) хранении:
усеченные формы. Гидролитические ферменты и химические вещества могут катализировать разрыв пептидных связей, что можно обнаружить с помощью ВЭЖХ или электрофореза в ПААГ с ДСН. В зависимости от свойств вариантов целевого продукта возможно использование пептидного картирования;
прочие модифицированные формы. Можно выявить дезаминированные, изомеризованные, с нарушенной дисульфидной связью, окисленные или измененные формы конъюгатов (гликозилирование, фосфорилирование) и охарактеризовать их с помощью хроматографических, электрофоретических и (или) других подходящих аналитических методов (например, высокоэффективной жидкостной хроматографии, капиллярного электрофореза, масс-спектроскопии, кругового дихроизма);
агрегаты. К агрегатам относятся димеры и полимеры целевого продукта. Их, как правило, не причисляют к целевому продукту и родственным соединениям и определяют их содержание с помощью подходящих аналитических методик (например, эксклюзионной хроматографии, капиллярного электрофореза).
Глава 7. Примеси ДНК и белков клетки-хозяина, стандартные
испытания в сравнении с валидационными исследованиями
1. Область применения
Биотехнологические препараты, преимущественно рекомбинантные белки, получают с помощью сложной системы экспрессии (производства) с использованием генетически модифицированных клеток (клеток бактерий, дрожжей или млекопитающих). Среди примесей, подлежащих удалению в процессе очистки на конечном этапе производства, 2 компонентами, представляющими наибольший интерес с точки зрения безопасности и переносимости, являются остаточная ДНК клетки-продуцента и остаточные белки клетки-хозяина. С момента регистрации первых рекомбинантных белков применяются разные подходы, основанные преимущественно на разновидностях систем производства, для работы с данными примесями. В качестве стандартного испытания на белки клетки-хозяина независимо от типа получаемого препарата и используемой клеточной системы необходимо определять остаточные белки клетки-хозяина, в то время как исследования остаточной ДНК в качестве стандартного испытания проводят только для препаратов, полученных из перевиваемых клеточных линий млекопитающих.
Необходимо гарантировать, что содержание таких примесей в лекарственном препарате, предназначенном для пациента, снижено до допустимого уровня. Для достижения этой цели следует рассмотреть два подхода:
валидационный подход: выполнить валидацию процесса производства для демонстрации того, что на заданных этапах процесса очистки эти примеси последовательно и воспроизводимо удаляются до тех пор, пока их уровень не станет приемлемым. Основываясь на коэффициентах снижения содержания примесей, можно предсказать и гарантировать остаточный уровень примеси в готовом препарате;
стандартный подход: разработать аналитические методики, позволяющие максимально точно контролировать уровень этих примесей на разных этапах процесса, и установить фиксированное предельное содержание примесей, что позволит надлежащим образом контролировать содержание таких примесей в готовом препарате.
Можно комбинировать эти два подхода, т.е. проводить стандартное испытание на раннем этапе процесса очистки, а валидационное испытание, демонстрирующее уменьшение содержания примесей, проводить на готовом препарате для выявления максимального содержания примесей.
Учитывая возможность применения рекомбинантных белков в международном масштабе, необходимо гармонизировать критерии оценки на международном уровне, чтобы производители могли разрабатывать свои препараты на одних и тех же производственных линиях, независимо от предполагаемых регионов регистрации и вывода препарата на рынок.
2. Остаточная ДНК клетки-продуцента
В отношении препаратов, полученных с помощью бактерий и дрожжей нет необходимости проводить стандартные испытания при условии, что в готовом препарате не превышены допустимые уровни остаточной ДНК, а в досье представлены достаточные данные по валидации. ДНК из стабильной клеточной линии млекопитающих ранее считалась фактором риска из-за опасений, что остаточная ДНК клетки-хозяина может обладать туморогенным потенциалом. Однако полученная в дальнейшем информация показала, что ДНК из стабильной клеточной линии представляет риск в гораздо меньшей степени, чем предполагалось раньше, и, следовательно, должна относиться к категории общих примесей.
Валидационные испытания (например, эксперименты по внесению заданных количеств примеси с соответствующим распределением фрагментов ДНК по размерам) должны проводиться с целью выявления основных этапов, на которых возможно снижение содержания ДНК, а также для занесения в документацию данных о способности данных этапов снижать содержание остаточной клеточной ДНК в готовом препарате до приемлемого и заданного уровней. В настоящее время методика количественного анализа ДНК подробно описана и обладает воспроизводимостью.
В дополнение к данным валидационных испытаний необходимо представить результаты количественного анализа ДНК для минимального количества производственных серий (например, 5 последовательных серий) с целью демонстрации воспроизводимости процесса производства в отношении снижения содержания остаточной ДНК до уровня, который предполагается получить в ходе валидационных испытаний. Учитывая удовлетворительные данные по валидации и достоверные результаты по ограниченному числу производственных серий, проведение стандартных испытаний для определения содержания ДНК в стабильной клеточной линии на этапе очистки нерасфасованного препарата (на других соответствующих этапах) представляется нецелесообразным.
В некоторых случаях (например, ранее не утвержденные перевиваемые клеточные линии, трансформация последовательностей ДНК вирусными векторами и т.д.) может возникнуть необходимость в применении стандартного контроля удаления ДНК.
Допустимо прекращение проведения стандартных испытаний ДНК для препаратов, выделенных из стабильных клеточных линий и для которых уже получено регистрационное удостоверение. Однако в таком случае необходимо внести официальные изменения в регистрационное досье. Документация с изменениями должна содержать удовлетворительные результаты валидационного испытания наряду с ретроспективными данными, полученными заявителем с момента начала производственного процесса, что подтвердит стабильный уровень содержания остаточной ДНК в ходе процесса.
3. Остаточные белки клетки-хозяина
Вопрос об остаточных белках клетки-хозяина затрагивает все системы экспрессии (производства), а не только экспрессионную систему с использованием клеток млекопитающих. Наличие в любых рекомбинантных белках таких примесей требует изучения потенциальной иммуногенности препарата. По этой причине в настоящее время необходимо контролировать содержание остаточных белков клетки-хозяина на этапе очистки нерасфасованного препарата с помощью подходящих методов анализа. Результаты испытаний от серии к серии должны соответствовать друг другу и находиться в установленных спецификацией пределах.
При определении допустимых пределов необходимо помнить, что невозможно установить общее предельное содержание остаточных белков клетки-хозяина для всех биотехнологических препаратов. Действительно, белки клетки-хозяина - примеси, качественная и количественная характеристика которых изменяется от одного препарата к другому и даже от одной системы производства (очистки) к другой. Стандартизация аналитических методов затруднена, поскольку выбор реагентов, используемых в ходе испытаний, зависит от получаемого препарата и системы экспрессии. В отношении остаточных белков клетки-хозяина трудно подобрать материал, обладающий схожими с примесями характеристиками, для использования на определенных этапах процесса или при использовании валидационного подхода (искусственно контаминированный материал).
В большинстве случаев при удалении белков используют хроматографические колонки, для которых избирательность (селективность) и выход методик зависят не только от качества материала, но также и от способа первичного и повторного использования колонок, условий хранения, санитарной обработки и периода их эксплуатации. Валидационный подход не может охватить все эти критические параметры.
В отношении остаточных белков клетки-хозяина валидационный подход применяют только для каждого препарата индивидуально с учетом:
качества аналитической методики, используемой для определения и количественного анализа примесей остаточных белков клетки хозяина;
дизайна и качества валидационных испытаний;
предполагаемого использования препарата (доза, показания, длительность лечения и т.д.).
4. Вывод
В большинстве случаев валидационный подход считается допустимым способом определения остаточного содержания ДНК клетки-хозяина.
Этот подход не так широко используется в отношении таких примесей, как белки клетки-хозяина, при анализе которых предпочтительно использовать индивидуальный подход для каждого препарата.
Необходимо отметить, что в любом случае нужно периодически проводить оценку процессов и методик анализа для подтверждения того, что при их использовании возможно достичь запланированных результатов.
Глава 8. Исследование стабильности биотехнологических
(биологических) препаратов
1. Введение
Указания, содержащиеся в актах, входящих в право Союза, регламентирующие изучение стабильности лекарственных средств, применяются в целом к биотехнологическим (биологическим) лекарственным препаратам. Тем не менее, биотехнологические (биологические) лекарственные препараты различаются по ряду характеристик, которые необходимо учитывать при составлении программы испытаний, направленной на подтверждение стабильности в течение планируемого срока годности. Для лекарственных препаратов, активными фармацевтическими субстанциями которых обычно являются белки и или полипептиды, поддержание молекулярной конформации, а следовательно, и биологической активности, обусловлено как нековалентными, так и ковалентными связями в молекулах. Эти препараты особенно чувствительны к окружающим факторам, например, к температурным изменениям, окислению, действию света, ионному составу среды и механическим воздействиям. Для сохранения их биологической активности и предотвращения их деградации необходимо соблюдение строгих условий хранения.
Для оценки стабильности могут потребоваться сложные аналитические методики. Испытания на биологическую активность (если их проведение возможно) должны быть частью основных исследований стабильности. Программа оценки стабильности также должна включать в себя соответствующие физико-химические, биохимические и иммунохимические методы анализа молекулярной сущности, а также методы количественного определения продуктов деградации, если чистота и молекулярные характеристики изучаемого препарата позволяют использовать эти методики.
Учитывая все указанное, заявитель должен получить надлежащие данные по стабильности биотехнологического (биологического) лекарственного препарата, учитывающие разнообразные условия окружающей среды, способные повлиять на активность, чистоту и качество. Основные данные, подтверждающие заявляемый срок годности как активной фармацевтической субстанции, так и лекарственного препарата, должны быть основаны на долгосрочных исследованиях стабильности (исследованиях естественного хранения) в реальном времени и реальных условиях. Таким образом, разработка надлежащей программы естественного хранения является критически важным этапом успешной разработки лекарственного препарата. Задача настоящей главы - представить заявителям рекомендации по видам исследований стабильности, результаты которых следует включить в регистрационное досье биологических лекарственных препаратов. В процессе экспертизы полученных сведений первичные данные по стабильности могут обновляться.
2. Область применения
Изложенные в настоящей главе положения распространяются на хорошо охарактеризованные белки и полипептиды, их производные и препараты, в состав которых они входят. Такие белки и полипептиды должны быть выделены из тканей, жидких сред организма, культур клеток или произведены с использованием технологии рекомбинантной ДНК. Таким образом, в настоящей главе описывается получение данных и представление результатов изучения стабильности таких биотехнологических (биологических) препаратов, как цитокины (интерфероны, интерлейкины, колониестимулирующие факторы, факторы некроза опухоли), эритропоэтины, активаторы плазминогена, плазменные факторы свертывания крови, гормоны роста и факторы роста, инсулины, моноклональные антитела и вакцины, состоящие из хорошо охарактеризованных белков или полипептидов. Кроме того, представленные ниже требования могут распространяться на другие типы лекарственных препаратов, например, традиционные вакцины, если это указано в специальных главах настоящих Правил или актах, входящих в право Союза. Рекомендации настоящей главы не распространяются на антибиотики, экстракты аллергенов, гепарины, витамины, цельную кровь или клеточные компоненты крови.
3. Терминология
Для целей настоящей главы используются понятия (термины), в значениях, установленных актами, входящими в право Союза и регламентирующими изучение стабильности лекарственных препаратов. Поскольку производители биотехнологических (биологических) препаратов используют также традиционную терминологию, в скобках приведены соответствующие традиционные термины.
4. Выбор серий
4.1. Активная фармацевтическая субстанция
(нерасфасованный материал)
При необходимости хранения нерасфасованного материала после его наработки до изготовления лекарственных форм и заключительных этапов производства требуется представить данные о стабильности по меньшей мере 3 серий, условия производства и хранения которых отражают условия промышленного масштаба. При необходимости хранения более 6 месяцев следует в первичное регистрационное досье включить результаты испытаний стабильности продолжительностью не менее 6 месяцев. Для активных фармацевтических субстанций с периодами хранения менее 6 месяцев минимальный объем данных по стабильности на момент первоначальной подачи регистрационного досье определяется в индивидуальном порядке. При подаче регистрационного досье допускается представление данных по опытно-промышленным сериям активной фармацевтической субстанции, произведенным с уменьшенным масштабом ферментации и очистки, при этом заявитель должен взять на себя обязательство о включении в программу долгосрочной оценки стабильности первых трех серий промышленного масштаба после получения регистрационного удостоверения.
Качество серий активной фармацевтической субстанции, предназначенных для программы изучения стабильности, должно отражать качество материала, изученного в доклинических и клинических исследованиях, а также качество материала, который будет производиться в промышленном масштабе. Кроме того, активная фармацевтическая субстанция (нерасфасованный материал), полученная в рамках опытно-промышленного производства, должна быть произведена с использованием процесса и условий хранения, отражающих условия промышленного масштаба. Активная фармацевтическая субстанция, включенная в программу изучения стабильности, должна храниться в контейнерах (упаковке), соответствующих по свойствам контейнерам (упаковке), которые будут использоваться для хранения при промышленном производстве. При изучении стабильности активной фармацевтической субстанции допускается использование контейнеров меньшего объема, если они произведены из аналогичного материала и с помощью той же системы "контейнер - укупорка", которые будут использоваться в промышленном производстве.
4.2. Промежуточные продукты
В процессе производства биотехнологических (биологических) препаратов качество и контроль качества определенных промежуточных продуктов могут быть критически важны для получения лекарственного препарата. В целом производитель должен идентифицировать промежуточные продукты и разработать собственные критерии и производственные ограничения, обеспечивающие их стабильность в рамках разработанного процесса. Несмотря на допустимость использования данных, полученных в опытно-промышленном масштабе, производитель должен подтвердить возможность экстраполяции полученных результатов на промышленный масштаб.
4.3. Лекарственный препарат (готовая лекарственная форма
в первичной упаковке)
Данные по стабильности следует представить, по меньшей мере, для 3 серий лекарственного препарата в первичной упаковке, соответствующего по свойствам тому препарату, который будет производиться в промышленном масштабе. По возможности серии лекарственного препарата в первичной упаковке, отобранные для изучения стабильности, необходимо произвести из разных серий активной фармацевтической субстанции. Если при регистрации заявленный срок годности превышает 6 месяцев, в регистрационное досье необходимо включить результаты изучения стабильности в течение не менее 6 месяцев. Для лекарственных препаратов со сроком годности менее 6 месяцев минимальный объем данных по стабильности при первичной подаче регистрационного досье определяется в индивидуальном порядке. Дата истечения срока годности лекарственного препарата будет установлена на основании фактических сведений, содержащихся в регистрационном досье. Поскольку срок годности препарата устанавливают на основании исследований, проведенных в реальном времени и при реальной температуре, в процессе экспертизы следует обновлять первоначальные данные по стабильности. Качество препарата в первичной упаковке, включенного в исследования стабильности, должно соответствовать качеству материала, изученного в доклинических и клинических исследованиях. На момент подачи регистрационного досье могут быть представлены данные по сериям лекарственного препарата, полученным в опытно-промышленных масштабах, при этом заявитель должен взять на себя обязательство по выполнению программы изучения долгосрочной стабильности 3 первых серий промышленного масштаба после получения регистрационного удостоверения. Если для установления срока годности препарата были представлены результаты испытания опытно-промышленных серий и препарат, произведенный в промышленном масштабе, не выдерживает спецификации на долгосрочную стабильность, составленную по результатам хранения опытно-промышленных серий, или не отражает свойства материала, изученного в доклинических и клинических исследованиях, заявитель в целях определения дальнейшей тактики должен уведомить об этом уполномоченные органы.
4.4. Отбор проб
Если один препарат реализуется в сериях, различающихся по номинальному объему (например, 1 мл, 2 мл или 10 мл), дозировке (например, 10, 20 и 50 ЕД) или массе (например, 1 мг, 2 мг или 5 мг), пробы для включения в программу оценки стабильности могут быть выбраны на основе матричного метода и (или) в исследовании крайних вариантов (брекетинг).
Матричный метод - статистический план изучения стабильности, при котором в определенный момент времени исследуется лишь подгруппа из общего числа проб всех комбинаций факторов, подлежащих испытанию. Его допускается использовать лишь в том случае, если представлено должное обоснование, подтверждающее, что стабильность испытуемых проб отражает стабильность всех проб. К различиям проб одного и того же препарата относится, например, охват различных серий, дозировок, размеров одной и той же системы "контейнер - укупорка" и, возможно, материалов системы "контейнер - укупорка". Если отсутствует обоснование, подтверждающее, что серии в одинаковых условиях ведут себя сходным образом, не допускается применять матричный метод к пробам, различия между которыми могут повлиять на стабильность, например, к различным дозировкам и контейнерам.
Если для трех номинальных объемов или более используется одинаковая дозировка и та же система "контейнер - укупорка", производитель вправе включить в программу лишь наименьший и наибольший размеры контейнера, то есть провести исследование крайних вариантов. План исследования крайних вариантов предполагает, что стабильность крайних вариантов отражает стабильность промежуточных вариантов. В некоторых случаях необходимо представить данные, подтверждающие, что полученные данные отражают свойства всех проб.
5. Профиль, свидетельствующий о стабильности
Универсальной методики или универсального перечня показателей, отражающего стабильность биотехнологического (биологического) средства, не существует. В связи с этим производитель должен разработать перечень показателей стабильности, обеспечивающий обнаружение изменений подлинности, чистоты и активности препарата.
На момент подачи регистрационного досье заявители должны располагать валидированными методами, составляющими профиль, свидетельствующий о стабильности, и данными для целей экспертизы. Перечень испытаний зависит от свойств препарата. Описанный ниже перечень не является исчерпывающим, но он отражает характеристики препарата, которые, как правило, необходимо документировать в целях надлежащего подтверждения его стабильности.
5.1. Протокол
В состав досье на регистрацию необходимо включить подробный протокол изучения стабильности как активной фармацевтической субстанции, так и лекарственного препарата. На основе этого протокола будут установлены условия хранения и срок годности. Протокол должен включать в себя всю необходимую информацию, подтверждающую стабильность биотехнологического (биологического) препарата в течение всего предлагаемого срока годности, включая, например, должным образом составленные спецификации и интервалы испытаний. Статистические методы, которые следует применять при подготовке документации, должны соответствовать требованиям актов, входящих в право Союза и регламентирующих изучение стабильности лекарственных средств.
5.2. Активность
Если предусмотренное применение лекарственного препарата связано с охарактеризованной и определяемой биологической активностью, испытание на активность должно стать частью исследований стабильности. В целях испытания стабильности продуктов, указанных в настоящей главе, под активностью понимаются определенная способность или свойство препарата оказывать планируемый эффект. Испытание основано на измерении некоторого показателя качества препарата и определяется подходящим количественным методом. Активность биотехнологических (биологических) препаратов, в целом испытанная различными лабораториями, может быть сопоставлена, если она нормирована по надлежащему стандартному материалу. С этой целью в методику необходимо включить стандартный материал, напрямую или косвенно калиброванный по соответствующему национальному или международному стандартному материалу.
Испытания на активность необходимо проводить через определенные интервалы, указанные в протоколе исследования стабильности, а результаты представлять в единицах биологической активности, калиброванных по возможности по национальному или международному стандарту. Если национальные и международные стандартные образцы отсутствуют, результаты исследования допускается выражать в самостоятельно разработанных единицах с использованием хорошо охарактеризованного стандартного материала.
Активность некоторых биотехнологических (биологических) препаратов зависит от конъюгации активных фармацевтических субстанций с другим соединением или связывания с адъювантом. Диссоциацию активных фармацевтических субстанций от носителя, используемого в качестве конъюгата или адъюванта, необходимо изучить в исследованиях реального времени при реальной температуре (включая условия транспортировки). Оценить стабильность таких препаратов бывает затруднительно, поскольку в некоторых случаях испытания in vitro на биологическую активность и для установления физико-химических свойств непрактичны или дают неточные результаты. В целях преодоления недостатков испытаний in vitro необходимо разработать надлежащие стратегии (например, испытание продукта до конъюгации (связывания), исследование высвобождения активной фармацевтической субстанции из связи с другим соединением, методики in vivo) или использовать подходящее суррогатное испытание.
5.3. Характеристики чистоты и молекулярных свойств
В целях испытания стабильности препаратов, описанных в настоящей главе, чистота - понятие относительное. Ввиду влияния гликозилирования, дезаминирования и других гетерогенностей определить абсолютную чистоту биотехнологического (биологического) препарата крайне сложно. В связи с этим чистоту биотехнологического (биологического) препарата необходимо, как правило, определять с помощью не менее двух методов. Получаемые значения чистоты зависят от выбранного метода. В целях испытания стабильности при испытании на чистоту необходимо основывать подтверждение качества на методах обнаружения продуктов деградации.
По возможности необходимо документировать и представить отчеты о степени чистоты, а также о содержании в биотехнологическом (биологическом) препарате, включенном в исследование стабильности, отдельных продуктов деградации и их суммы. Допустимые предельные содержания продуктов деградации необходимо выработать на основании аналитического профиля серий активной фармацевтической субстанции и лекарственного препарата, использованных в доклинических и клинических исследованиях.
Используя соответствующую физико-химическую, биохимическую и иммунохимическую аналитическую методологию, необходимо всесторонне охарактеризовать свойства активной фармацевтической субстанции и (или) лекарственного препарата (например, молекулярную массу, заряд, гидрофобность), а также точно установить изменения, обусловленные деградацией, вследствие дезаминирования, окисления, сульфоксилирования, агрегации или фрагментации при хранении. Примерами таких методов являются электрофорез (электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (ДСН-ПААГ), иммуноэлектрофорез, вестерн-блот, изоэлектрическое фокусирование), хроматография с высокой разрешающей способностью (например, обращенно-фазная хроматография, гель-фильтрация, ионный обмен, аффинная хроматография) и пептидное картирование.
Если по результатам естественного, ускоренного хранения и (или) стресс-исследований стабильности обнаруживаются значимые качественные или количественные изменения, свидетельствующие об образовании продукта деградации, следует проанализировать его потенциальную опасность и оценить необходимость установления характеристик продуктов деградации и их количественного определения в рамках программы естественного хранения. Необходимо представить и обосновать допустимые пределы, учитывая содержание продуктов деградации в материале, использованном в доклинических и клинических исследованиях.
В отношении веществ, свойства которых охарактеризовать надлежащим образом невозможно, и лекарственных препаратов, чистоту которых невозможно определить с помощью стандартных аналитических методов, заявитель обязан предложить альтернативные аналитические методики и обосновать их.
5.4. Прочие характеристики препаратов
Необходимо отслеживать и представлять результаты испытаний следующих характеристик (которые не являются присущими исключительно биотехнологическим (биологическим) препаратам) лекарственного препарата, заключенного в окончательную первичную упаковку: внешний вид (цвет и мутность раствора (суспензии); цвет, консистенция и время растворения порошков), видимые включения в растворах или после восстановления порошков и лиофилизатов, pH и влажность порошков и лиофилизатов.
Необходимо по меньшей мере в начале и конце предлагаемого срока годности провести испытания на стерильность или альтернативные испытания (например, испытание целостности системы "контейнер - укупорка").
В течение срока годности лекарственного препарата добавки (например, стабилизаторы, консерванты) и другие вспомогательные вещества могут подвергаться деградации. Если по результатам предварительных исследований стабильности обнаруживаются признаки реакции или деградации материалов, негативно влияющих на качество лекарственного препарата, может возникнуть необходимость контроля указанных показателей в рамках программы изучения стабильности.
Система "контейнер - укупорка" может негативно сказаться на качестве продукта и требует тщательного оценки (как это указано ниже).
6. Условия хранения
6.1. Температура
Поскольку большинство готовых биотехнологических (биологических) лекарственных препаратов подлежит хранению при строго заданной температуре, условия хранения в исследованиях стабильности в реальном времени при реальной температуре могут быть ограничены такой температурой хранения.
6.2. Влажность
Биотехнологические (биологические) препараты, как правило, выпускаются в контейнерах, защищающих их от влаги. В связи с этим, если подтверждено, что предлагаемый контейнер (и условия хранения) обеспечивает достаточную защиту от высокой и низкой влажности, изучение стабильности при различной относительной влажности, как правило, не требуется. Если влагоустойчивые контейнеры не используются, необходимо представить соответствующие данные по стабильности.
6.3. Ускоренные и стресс-условия
Срок годности необходимо определять на основании исследований в реальном времени при реальной температуре. В то же время настоятельно рекомендуется проведение исследований активной фармацевтической субстанции и лекарственного препарата в ускоренных и стресс-условиях. При определении срока годности результаты ускоренного хранения могут послужить источником ценных вспомогательных данных для установления даты истечения срока годности, служить источником данных по стабильности для целей дальнейшей разработки (например, предварительной оценки предлагаемых изменений процесса производства, таких как изменение состава (формуляции), укрупнение), содействия в валидации аналитических методик для использования в программе изучения стабильности и наработки данных, позволяющих установить профиль деградации активной фармацевтической субстанции или лекарственного препарата. Исследования в стресс-условиях позволяют определить, насколько пагубно влияет на лекарственный препарат случайное воздействие условий, отличных от предлагаемых для хранения (например, при транспортировке), а также выявить определенные испытуемые показатели качества, наилучшим образом отражающие стабильность препарата. Испытания активной фармацевтической субстанции и лекарственного препарата, помещенных в экстремальные условия, могут способствовать выявлению механизмов деградации; если таковая обнаруживается, то необходимо осуществлять контроль возможных изменений при хранении в предлагаемых условиях. Несмотря на то что в актах, входящих в право Союза, и регламентирующих изучение стабильности лекарственных средств, описаны требования к исследованию в ускоренных и стресс-условиях, заявителю необходимо учитывать, что они могут не подходить для биотехнологических (биологических) лекарственных препаратов. В связи с этим условия необходимо тщательно подбирать в индивидуальном порядке.
6.4. Свет
За получением рекомендаций по проведению испытаний заявителям необходимо обращаться в соответствующие уполномоченные органы в индивидуальном порядке.
6.5. Система "контейнер - укупорка"
Ввиду взаимодействия биотехнологического (биологического) лекарственного препарата с его системой "контейнер - укупорка" возможно изменение его качества. Поскольку для жидких лекарственных препаратов такое взаимодействие не исключается (за исключением запаянных ампул), в целях определения влияния укупорки на качество лекарственного препарата в исследование стабильности необходимо включить пробы, которые следует расположить в перевернутом или горизонтальном положении (то есть в контакте с укупоркой), а также в вертикальном положении. Необходимо представить данные для всех возможных комбинаций систем "контейнер - укупорка", которые предполагается выпускать на рынок Союза.
В дополнение к стандартным данным, необходимым для обычного флакона для однократного использования, заявитель должен подтвердить, что укупорка, используемая во флаконе, содержащем несколько доз, способна выдержать условия многократных введений и извлечений, обеспечивая сохранение полной активности, чистоты и качества лекарственного препарата на максимальный срок, указанный в инструкциях по применению на контейнерах, упаковках и (или) в листках-вкладышах. Такая информация о лекарственном препарате должна соответствовать требованиям к инструкции по медицинскому применению лекарственных средств и общей характеристике лекарственного препарата для медицинского применения Союза, утверждаемым Комиссией.
6.6. Стабильность восстановленного лиофилизированного
лекарственного препарата
Необходимо подтвердить стабильность лиофилизированных лекарственных препаратов после их восстановления в условиях и при максимальном периоде хранения, указанных в инструкциях по использованию на контейнерах, упаковках и (или) в листках-вкладышах. Такая информация о лекарственном препарате должна соответствовать Требованиям к инструкции по медицинскому применению лекарственных средств и общей характеристике лекарственного препарата для медицинского применения Союза.
7. Частота испытаний
Срок годности биотехнологических (биологических) лекарственных препаратов составляет от нескольких дней до нескольких лет. В связи с этим сложно составить универсальные рекомендации по продолжительности исследования стабильности и частоте испытаний, которые были бы справедливы для всех видов биотехнологических (биологических) лекарственных препаратов. Тем не менее за редким исключением сроки годности одобренных и потенциальных будущих лекарственных препаратов попадают в диапазон от полугода до 5 лет. Поэтому указанные ниже принципы ориентированы на срок годности, укладывающийся в этот диапазон. Данный подход учитывает тот факт, что деградация биотехнологических (биологических) лекарственных препаратов может протекать под влиянием одних и тех же факторов в течение различных интервалов долгосрочного хранения.
Если предлагаемый срок годности менее 1 года, исследования стабильности в реальных условиях в первые 3 месяца необходимо проводить ежемесячно, далее - каждые 3 месяца.
Если предлагаемый срок годности более 1 года, то исследования необходимо проводить каждые 3 месяца в течение первого года, каждые 6 месяцев - в течение второго года, далее - ежегодно.
Несмотря на то что указанные выше интервалы испытаний подходят для предрегистрационного этапа, при наличии данных, подтверждающих требуемую стабильность, после получения регистрационного удостоверения допускается сократить частоту испытаний. Если данные подтверждают, что стабильность лекарственного препарата не снижается, заявителю рекомендуется представить протокол, обосновывающий исключение определенных интервалов испытаний (например, на сроке 9 месяцев) в рамках пострегистрационных долгосрочных исследований.
8. Спецификации
Несмотря на то что биотехнологические (биологические) лекарственные препараты могут претерпевать значительное снижение активности, физико-химические изменения или деградацию при хранении, в международных и национальных правилах содержится недостаточно рекомендаций по составлению различающихся спецификаций на выпуск и конец срока годности (обращение). Рекомендации по максимально допустимому снижению активности, предельным физико-химическим изменениям и деградации в рамках предлагаемого срока годности для отдельных видов и групп биотехнологических (биологических) лекарственных препаратов не составлялись, поэтому их рассматривают в индивидуальном порядке. Каждый продукт должен соответствовать своим спецификациям в пределах, установленных для безопасности, чистоты и активности на протяжении всего предлагаемого срока годности. Указанные спецификации и пределы необходимо устанавливать на основании всей доступной информации, используя необходимые статистические методы. Использование различающихся спецификаций на выпуск и конец срока годности (обращение) необходимо обосновать достаточным объемом данных, подтверждающих, что клинические свойства не ухудшаются, как указано в требованиях актов, входящих в право Союза и регламентирующих изучение стабильности лекарственных средств.
9. Информация о препарате
Большинство биотехнологических (биологических) активных фармацевтических субстанций и лекарственных препаратов рекомендуется хранить при строго заданных температурах. Необходимо предусмотреть специальные указания, особенно для активных фармацевтических субстанций и лекарственных препаратов, не выдерживающих замораживание. Такие условия и, в соответствующих случаях, рекомендации по защите от света и (или) влажности, необходимо указывать на контейнерах, упаковках и (или) в инструкциях по медицинскому применению (листках-вкладышах). Такая информация о препарате должна соответствовать требованиям к инструкции по медицинскому применению лекарственных средств и общей характеристике лекарственного препарата для медицинского применения, утверждаемым Комиссией.
10. Определения
Для целей настоящей главы используются понятия (термины), которые означают следующее:
"конъюгат" - состоит из активной фармацевтической субстанции (например, пептида или углевода), ковалентно или нековалентно связанного с носителем (например, белком, пептидом или неорганическим минералом) в целях улучшения эффективности или стабильности лекарственного препарата;
"опытно-промышленное производство" - производство активной фармацевтической субстанции или лекарственного препарата с помощью процедуры, полностью отражающей и повторяющей таковую при промышленном производстве. Методы культивирования клеток, сбора и очистки должны совпадать, за исключением масштаба производства;
"примесь" - любой компонент активной фармацевтической субстанции или лекарственного препарата, который не является активной фармацевтической субстанцией, вспомогательным веществом или иной добавкой лекарственного препарата;
"продукт деградации" - молекула, образующаяся вследствие изменения со временем активной фармацевтической субстанции. В целях испытания стабильности препаратов, указанных в настоящей главе, такие изменения могут возникать вследствие обработки или хранения (например, при дезаминировании, окислении, агрегации или протеолиза). Некоторые продукты деградации биотехнологических (биологических) препаратов могут обладать активностью;
"промежуточный продукт" - в отношении биотехнологического (биологического) препарата - материал, получаемый в ходе процесса производства, который не является активной фармацевтической субстанцией или лекарственным препаратом, но производство которого необходимо для успешного получения активной фармацевтической субстанции или лекарственного препарата. Промежуточный продукт в целом поддается количественному определению и для него разрабатывается спецификация, позволяющая до продолжения производственного процесса определить успешность предыдущего этапа производства. К ним относятся материалы, которые могут подвергаться дальнейшей молекулярной модификации или храниться в течение определенного времени до дальнейшей обработки;
"промышленное производство - производство в масштабе, как правило, на оборудовании, предназначенном для производства лекарственного препарата, выпускаемого на рынок Союза.
Глава 9.1. Сопоставимость показателей качества
биотехнологических (биологических) препаратов при внесении
изменений в производственный процесс
1. Введение
1.1. Цели
В настоящей главе представлены основные принципы оценки сопоставимости биотехнологических (биологических) продуктов (под которыми подразумевается промежуточный продукт, фармацевтическая субстанция и лекарственный препарат), полученных до и после внесения изменений в производственный процесс (под которым подразумеваются производственный процесс, производственные мощности (помещения) и оборудование, которые могут повлиять на критические параметры обработки и, таким образом, на качество) получения действующего вещества или готовой формы лекарственного препарата. Настоящая глава содержит указания по сбору информации, необходимой для подтверждения того, что изменения производственного процесса не окажут негативного влияния на показатели качества, безопасности и эффективности биотехнологического (биологического) продукта. Глава не предписывает конкретные аналитические, доклинические и клинические стратегии, основное внимание уделено вопросам качества.
1.2. Общие указания
Производители (в том числе третья сторона, имеющая соглашение (договор) на производство промежуточных продуктов, активной фармацевтической субстанции или лекарственного препарата от имени держателя регистрационного удостоверения (разработчика - если лекарственный препарат не зарегистрирован)) биотехнологических (биологических) продуктов как правило в процессе разработки и после регистрации лекарственного препарата вносят изменения в производственный процесс биотехнологического (биологического) продукта. Изменения в производственный процесс вносятся с целью совершенствования технологического процесса, масштабирования производства, повышения стабильности продукта и при изменении регуляторных требований к производственному процессу. При внесении изменений в производственный процесс производитель, как правило, должен оценить соответствующие показатели качества и подтвердить, что вносимые изменения не оказывают негативного влияния на показатели безопасности и эффективности продукта. Если результаты исследований сопоставимости свидетельствуют о том, что улучшенные качества приводит к значимому повышению эффективности и (или) безопасности, препарат до и после изменений может оказаться несопоставимым, однако такие результаты могут рассматриваться как приемлемые. Производителям рекомендуется обратиться, в данном случае, за консультацией в уполномоченный орган (организацию). На основании данных исследований делается заключение о необходимости проведения подтверждающих доклинических и клинических исследований.
1.3. Сфера применения
Настоящая глава, взаимосвязана с другими главами настоящих Правил и содержит описание дополнительных подходов, посвященных:
сравнению продукта до и после внесения изменения в производственный процесс,
оценке влияния изучаемых различий по показателям качества, обусловленных изменением технологического процесса продукта, на показатели безопасности и эффективности продукта.
Представленные в настоящей главе требования распространяются на все нижеуказанные продукты и процессы:
белки и полипептиды, их производные, а также продукты, в которых они являются компонентами, например, конъюгаты. Белки и полипептиды могут быть получены с помощью рекомбинантных и нерекомбинантных экспрессирующих систем, хорошо поддаются очистке и установлению характеристик с использованием соответствующего набора аналитических методик;
производственный процесс, изменения в котором сделаны одним производителем (включая контрактных производителей), который может прямо провести сравнение результатов испытаний продукта, полученных до и после внесения изменения в производственный процесс;
продукты, которые находятся в процессе разработки, и зарегистрированные лекарственные препараты.
Указанные в настоящей главе требования применимы к другим типам биологических лекарственных препаратов, например, белкам или полипептидам, выделенным из тканей и жидкостей организма. При этом производителю рекомендуется проконсультироваться с уполномоченным органом государства-члена.
1.4. Основные принципы
Целью исследований сопоставимости является обеспечение качества, безопасности и эффективности продукта, полученного с измененным процессом производства, за счет сбора и анализа соответствующих данных, направленных на выявление любого неблагоприятного влияния на продукт, обусловленного внесенными изменениями.
Подтверждение сопоставимости необязательно должно выражаться в идентичности показателей качества продукта до и после внесения изменений, однако они должны быть высоко сопоставимы, а текущие знания должны позволять провести достаточное прогнозирование возможности обеспечения отсутствия нежелательного влияния на безопасность и эффективность лекарственного препарата.
Определение сопоставимости основывается на результатах аналитических и биологических испытаний и в некоторых случаях на данных доклинических и клинических исследований. Если производителем представлены убедительные доказательства сопоставимости на основании аналитических исследований, проведение доклинических и клинических исследований с продуктом, полученным после внесения изменений, не требуется. Однако если зависимость безопасности и эффективности от отдельных показателей качества не установлена и выявлены различия в показателях качества продукта до и после внесения изменений, в программу подтверждения сопоставимости необходимо включить совокупность сравнительных испытаний качества, доклинических и (или) клинических исследований.
Для оценки влияния изменений производственного процесса необходимо провести анализ всех потенциальных последствий для продукта. С учетом данной оценки необходимо установить критерии определения высокой сопоставимости измененного продукта. Проводится сбор и анализ всех данных, касающихся продуктов, полученных до и после внесения изменений, в том числе таких как результаты рутинного контроля качества серий, внутрипроизводственного контроля, валидации (оценки) процесса производства, установления характеристик и оценки стабильности (если применимо). Сравнение полученных результатов с заранее определенными критериями должно позволить получить объективную оценку сопоставимости продуктов, полученных до и после внесения изменений в производственный процесс.
Результаты сравнительной оценки показателей качества позволяют сделать один из следующих выводов:
продукты, полученные до и после изменения высоко сопоставимы по показателям качества, то есть отрицательного влияния на профили безопасности и эффективности не предвидится;
продукты, полученные до и после внесения изменений в производственный процесс, высоко сопоставимы, но используемых для сравнения аналитических методик недостаточно, чтобы выявить различия, которые могут повлиять на безопасность и эффективность продукта. Для получения окончательного вывода производитель должен рассмотреть вопрос о проведении дополнительных исследований (например, дополнительная оценка по показателям качества) или доклинических и клинических исследований;
продукты, полученные до и после внесения изменений, высоко сопоставимы между собой, однако выявляются некоторые различия между ними по показателям качества. При этом представлено обоснование (на основании накопленного опыта, соответствующей информации и данных) того, что данные различия не оказывают отрицательного влияния на безопасность и эффективность продукта. В данной ситуации продукты, полученные до и после внесения изменений, сопоставимы;
продукты, полученные до и после внесения изменений, сопоставимы между собой, но выявлены некоторые различия между ними по показателям качества и имеется опасение о неблагоприятном влиянии изменений на профили безопасности и эффективности. В данной ситуации дополнительный сбор информации по показателям качества не позволяет получить положительное заключение о сопоставимости. Необходимо рассмотреть вопрос о проведении доклинических и клинических исследований;
показатели качества исследуемых продуктов различаются значительно, это не позволяет признать продукты до и после внесения изменений высоко сопоставимыми. Требования, изложенные в настоящей главе, на такие случаи не распространяются.
2. Основные требования
2.1. Принципы исследования сопоставимости
Основной целью исследований сопоставимости является подтверждение высокой сопоставимости продуктов, полученных до и после внесения изменений, по показателям качества, безопасности и эффективности. Для достижения данной цели продукт должен быть изучен на той стадии процесса, которая является наиболее подходящей для обнаружения изменения показателей качества. Поэтому может потребоваться проведение исследований на разных стадиях производства. Например, если изменение затронуло процесс производства активной фармацевтической субстанции и могло повлиять на качество лекарственного препарата, для оценки сопоставимости целесообразно собрать данные как об активной фармацевтической субстанции, так и о лекарственном препарате. Сопоставимость может быть установлена на основании испытаний показателей качества (ограниченных или полных), но иногда могут потребоваться дополнительные связующие исследования сопоставимости. Объем исследований сопоставимости зависит от следующих факторов:
стадия производства, на которой внесены изменения;
потенциальное влияние внесенных изменений на чистоту, физико-химические и биологические свойства с учетом сложности и степени изученности продукта (например, примеси, родственные соединения);
доступность аналитических методик, позволяющих обнаружить потенциальные изменения характеристик, и результаты этих исследований;
зависимость между показателями качества, безопасностью и эффективностью, исходя из доклинического и клинического опыта.
Для оценки сопоставимости необходимо провести анализ следующих данных (перечень не является исчерпывающим):
физико-химические и биологические свойства, полученные при установлении характеристик по показателям качества продукта;
результаты анализа проб, взятых на разных стадиях производства (промежуточный продукт, активная фармацевтическая субстанция, лекарственный препарат);
необходимость данных по стабильности, в том числе данных полученных по результатам ускоренного хранения или стресс-стабильности, чтобы охарактеризовать потенциальные различия между путями деградации продукта и, как следствие, потенциальные различия между родственными соединениями и родственными примесями;
серии, использованные для подтверждения постоянства производственного процесса;
ранее полученные данные, позволяющие охарактеризовать потенциальный дрейф (устойчивое во времени непрерывное изменение показателя, подтвержденное результатами статистического или графического анализа временного ряда) показателей качества с точки зрения безопасности и эффективности после единичного изменения либо серии изменений процесса производства. То есть производитель обязан изучить влияние ряда изменений во времени, чтобы подтвердить, что неприемлемого влияния на профили безопасности и эффективности не произошло.
При оценке полученных данных необходимо также учитывать следующее:
критические контрольные точки производственного процесса, неблагоприятно влияющие на характеристики продукта, например, влияние изменения процесса производства на качество внутрипроизводственных материалов, а также возможность использования материала, полученного после изменения процесса культивирования клеток, на последующих этапах;
адекватность внутрипроизводственного контроля, включая критические контрольные точки и внутрипроизводственные испытания: в целях поддержания качества продукта необходимо подтвердить, модифицировать или создать внутрипроизводственные контрольные точки измененного процесса производства;
доклинические и клинические характеристики лекарственного препарата, а также его показания к применению (в соответствии с требованиями подраздела 2.5 настоящей главы).
2.2. Вопросы качества
2.2.1. Аналитические подходы.
Аналитические методы при исследовании сопоставимости необходимо тщательно подобрать и оптимизировать по отношению к продукту для того, чтобы обеспечить максимальную возможность выявления значимых различий в показателях качества, являющихся следствием вносимых изменений в процесс производства. Для того чтобы достаточно полно оценить физико-химические, иммунохимические свойства и биологическую активность продукта, может потребоваться несколько аналитических методик для исследования одного и того же показателя (например, молекулярной массы, примесей, вторичной и (или) третичной структуры белка). В таких случаях методы должны быть основаны на разных физико-химических или биологических принципах, чтобы обеспечить максимальную возможность для выявления различий между продуктами, обусловленных изменением процесса производства.
В некоторых случаях в силу ограничений таких методик (например, прецизионности, специфичности и предела обнаружения) и сложности некоторых продуктов, обусловленной их молекулярной гетерогенностью, достаточно затруднительно обеспечить обнаружение модификации продукта с помощью набора аналитических методик, выбранного для такого продукта до внесения изменения.
Производитель должен установить:
пригодны ли существующие испытания для их целевого назначения или их необходимо модифицировать. Например, если изменение процесса производства приводит к образованию другого профиля белков клеток хозяина, производители обязаны подтвердить, что испытание, использованное для количественного определения таких примесей, все еще пригодно для его целевого назначения. В данном случае целесообразно модифицировать существующее испытание;
необходимость добавления новых испытаний вследствие изменений показателей качества, которые имеющиеся методики не способны определить. Таким образом, если при внесении изменений в производственный процесс ожидаются определенные изменения показателей качества (например, после использования нового сырья или изменения стадии хроматографической очистки), в целях установления характеристик или рутинных выпускающих испытаний целесообразно разработать новые аналитические методики, то есть предусмотреть дополнительные аналитические подходы, помимо ранее использованных подходов.
Определение показателей качества в исследованиях по установлению характеристик необязательно влечет использование валидированных методик, но такие методики должны быть научно обоснованными и давать достоверные результаты. Методики, использованные для определения показателей качества при выпуске серий, требуют валидации в соответствии с главами 6 и 8 настоящих Правил и требованиями по валидации аналитических методик актов, входящих в право Союза.
2.2.2. Установление характеристик.
В соответствии с главой 6 настоящих Правил установление характеристик биотехнологических (биологических) продуктов включает в себя определение физико-химических свойств, биологической активности, иммунохимических свойств (если применимо), чистоты, примесей, контаминантов и количественного содержания (quantity).
Если внесено изменение в процесс производства, влияющее на показатели качества, в целях прямого сравнения продукта до и после изменения, как правило, требуется полное или ограниченное (при наличии обоснований) повторение установления характеристик, проведенного при регистрации лекарственного препарата. Тем не менее в некоторых случаях может потребоваться дополнительное установление характеристик. Например, если изменения процесса производства приводят к получению профиля характеристик продукта, отличного от профиля, полученного по результатам доклинических и клинических исследований, или иного репрезентативного профиля (например, стандартные материалы, серии, находящиеся в обороте), необходимо изучить значимость таких изменений. Результаты всестороннего установления характеристик материала, использованного в опорных клинических исследованиях, могут представлять собой необходимую опорную точку для последующих исследований сопоставимости.
Каждый из указанных ниже критериев следует рассматривать в качестве ключевого фактора при проведении исследований сопоставимости:
физико-химические свойства. При планировании и проведении исследований сопоставимости производитель должен придерживаться концепции целевого продукта (и его вариантов), описанной в главе 6 настоящих Правил. Необходимо также учесть сложность молекулярной структуры и степень ее молекулярной гетерогенности. Следует убедиться в сохранности вторичной, третичной и четвертичной структур белка, полученного после внесения изменений в производственный процесс. Если подобную информацию о структуре получить невозможно, результаты соответствующих методов количественного определения биологической активности (как это указано далее) могут свидетельствовать о правильной конформационной структуре;
биологическая активность. При подтверждении показателей качества лекарственного средства, которые представляют ценность при установлении свойств и анализе серий, результаты определения биологической активности могут служить нескольким целям, а в некоторых случаях могут свидетельствовать о клинических эффектах. Производители должны учитывать ограничения, присущие биологическим испытаниям (например, высокая вариабельность, ограничения, способные воспрепятствовать обнаружению различий), которые могут возникать вследствие изменения процесса производства.
Если определение биологической активности дополняет результаты физико-химических исследований, например, исследование структуры белка более высокого порядка, использование соответствующего биологического испытания с достаточной прецизионностью и правильностью может служить косвенным подтверждением того, что при изменении производственного процесса не произошло изменений структур более высокого порядка. Если физико-химические или биологические испытания не позволяют убедиться в неизменности структуры более высокого порядка, целесообразно провести доклинические и (или) клинические исследования.
Если изменения вносятся в процесс производства продукта, проявляющего широкий спектр биологических активностей, следует определить набор функциональных испытаний для оценки всего спектра этих активностей. Например, определенные белки обладают несколькими функционально активными доменами, которые обусловливают проявление ферментативной и рецептор-опосредованной активности. В указанных случаях необходимо предусмотреть изучение всех значимых видов функциональной активности продукта.
Если один или более видов активности не полностью коррелируют с клинической безопасностью и эффективностью или механизм действия неясен, следует подтвердить, что доклинические и клинические эффекты продукта, полученные после изменения производственного процесса, не изменились;
иммунохимические свойства. Если иммунохимические свойства являются частью установления характеристик продукта (например, для антител или продуктов на их основе), следует подтвердить, что продукт, полученный после изменения процесса производства, сопоставим по указанным специфическим свойствам с неизмененным;
чистота, примеси и контаминанты. С помощью набора выбранных аналитических методик необходимо получить данные для оценки, того, произошло ли изменение профиля чистоты с точки зрения целевого продукта.
Если выявлены различия в профиле чистоты и примесей продуктов, должны быть проведены исследования по определению их влияния на безопасность и эффективность продукта, полученного после изменения процесса производства. Если изменение привело к появлению новых примесей, необходимо их идентифицировать и охарактеризовать (если это возможно). В зависимости от вида и количества примеси требуется проведение дополнительных доклинических и (или) клинических исследований с целью подтверждения отсутствия негативного влияния на профиль безопасности и эффективности лекарственного препарата. Отсутствие дополнительных исследований должно быть обосновано.
Используя удовлетворительные внутрипроизводственные критерии приемлемости или пределы действия для активной фармацевтической субстанции или лекарственного препарата, необходимо избегать наличия контаминантов и (или) надлежащим образом их контролировать. В целях определения влияния новых контаминантов на качество, безопасность и эффективность лекарственного средства необходимо их оценить.
2.2.3. Спецификации.
Методики, используемые для оценки показателей качества активной фармацевтической субстанции или лекарственного препарата, включенные в спецификацию, обычно недостаточны для оценки влияния изменений производственного процесса на качество продукта, так как они подобраны для рутинного подтверждения качества, а не для полной характеристики продукта. Следует подтвердить, что после изменения процесса производства спецификации продолжают обеспечивать контроль качества продукта. Результаты анализа, соответствующие требованиям спецификации, но выходящие за пределы предыдущих трендов производства, могут свидетельствовать о различиях между продуктами до и после изменений и требовать дополнительного исследования или анализа. Если данные указывают на то, что предыдущее испытание более не подходит для рутинного выпускающего контроля качества серий измененного продукта, может потребоваться модификация, исключение или добавление испытания в спецификацию. Например, исключение из среды для культивирования клеток бычьей сыворотки позволяет исключить и проведение соответствующих испытаний. Однако при отсутствии обоснований расширение критериев приемлемости, как правило, недопустимо. В отдельных случаях, если в результате изменения процесса производства меняется профиль примесей, могут потребоваться дополнительные испытания и новые критерии приемлемости. При оценке как аналитических методик, так и критериев приемлемости измененного продукта необходимо руководствоваться общими принципами составления спецификаций, указанными в главе 6 настоящих Правил, то есть влиянием изменений на валидированный процесс производства, результатами исследований по установлению характеристик, данными анализа серий, данными по стабильности и доклиническим и клиническим опытом.
2.2.4. Стабильность.
Даже незначительное изменение производственного процесса, может повлиять на стабильность продукта. Любые изменения, которые могут привести к изменениям структуры белка, показателей чистоты и профиля примесей, должны быть оценены в отношении их влияния на стабильность, поскольку белки часто чувствительны к таким изменениям, как состав буферного раствора, условия обработки и перерыва в производстве (holding), использование органических растворителей и т.д. С помощью исследований стабильности возможно обнаружение незначительных различий, которые невозможно обнаружить в исследованиях по установлению характеристик. Например, наличие следовых количеств протеазы может быть определено только по продуктам деградации, которые образуются через продолжительный период хранения продукта. В некоторых случаях двухвалентные ионы, вымываемые из укупорочной системы, могут изменить профиль стабильности вследствие активации следовых протеаз, не обнаруженных в исследованиях стабильности неизмененного лекарственного средства. Таким образом, в соответствующих случаях в отношении измененного лекарственного средства необходимо начать исследования стабильности в реальном времени и при реальной температуре.
Ускоренные и стрессовые исследования стабильности также являются полезным инструментом для определения возможной деградации белкового продукта и обеспечивают возможность прямого сравнения свойств продуктов, полученных до и после изменения технологического процесса. Полученные таким образом результаты могут свидетельствовать о различиях между свойствами продукта, требующих дополнительного изучения, а также помогают определить отклонения, указывающие на необходимость введения дополнительных контрольных мероприятий в процесс производства и хранения для устранения таких неожиданных различий в продукте. Необходимо провести соответствующие исследования, подтверждающие, что условия хранения и контрольные испытания выбраны правильно.
В целях определения условий проведения исследований стабильности, которые позволят получить необходимые данные для осуществления сравнения лекарственного средства до и после изменений, следует выполнять требования главы 8 настоящих Правил и регламентирующих изучение стабильности лекарственных средств актов, входящих в право Союза.
2.3. Производственный процесс.
Хорошо охарактеризованный процесс производства и контроль такого процесса производства обеспечивают получение продукта приемлемого качества на постоянной основе. Подходы к оценке влияния любых изменений производственного процесса зависят от особенностей процесса, самого продукта, знаний о процессе и опыта производителя, а также от данных, полученных при разработке продукта. Следует подтвердить, что контроль измененного процесса обеспечит сходный или более эффективный контроль качества продукта по сравнению с первоначальным процессом.
Необходимо обязательно провести тщательный анализ потенциального влияния планируемых изменений на последующие стадии производства и зависящие от них показатели качества (например, критерии приемлемости, внутрипроизводственные спецификации, внутрипроизводственные испытания, время хранения продукта между производственными стадиями, пределы действия, валидация (оценка)). Такой анализ позволяет выявить испытания, которые необходимо провести в ходе исследований сопоставимости, какие внутрипроизводственные критерии приемлемости или критерии приемлемости на выпуск серии либо аналитические методики требуют пересмотра, а также стадии, на которые не должны повлиять предлагаемые изменения. Например, анализ промежуточных продуктов может выявить потенциальные различия в продукте, что требует оценки пригодности использующихся аналитических методик для выявления таких различий. Решение об исключении из анализа некоторых этапов производственного процесса необходимо научно обосновать.
Если технологический процесс и связанные с ним методы контроля предполагается изменить, необходимо подтвердить, что продукты, полученные до и после внесения изменений, сопоставимы. Для подтверждения сопоставимости следует показать, что специфические промежуточные продукты сопоставимы или измененный процесс может обеспечивать удаление производственных и родственных примесей, включая новые примеси, которые образовались в результате изменения процесса. Если изменения вносятся в процесс производства зарегистрированных лекарственных препаратов, как правило, требуется сравнение с данными о результатах испытаний серий промышленного масштаба.
При анализе процесса производства необходимо учитывать такие факторы, как критичность стадии производственного процесса и предлагаемого изменения, место изменения в производственном процессе и потенциальное влияние изменения на остальные стадии процесса производства, а также характер и степень изменения. Информацию, которая может в этом помочь, можно получить из разных источников: сведения, полученные в исследованиях по разработке, исследованиях по валидации или оценке (при невозможности валидации) маломасштабных серий; опыт ранее проведенных изменений, сведения об изменении схожих процессов производства аналогичных лекарственных препаратов, данные литературы. Несмотря на то что сведения из внешних источников могут представлять определенную ценность, анализ изменения необходимо осуществлять в контексте конкретного процесса производства и конкретного продукта.
При внесении изменений в производственный процесс производитель должен подтвердить, что соответствующие контрольные мероприятия, включая новые, обеспечат получение сопоставимого лекарственного препарата. Измененные стадии процесса производства необходимо повторно оценить и (или) валидировать. Внутрипроизводственный контроль, включая ключевые контрольные точки и внутрипроизводственные испытания, должен обеспечить надлежащий контроль измененного процесса и поддерживать качество продукта. При отсутствии каких-либо свидетельств, что изменение влияет на функционирование последующих после культивирования стадий производства или качество промежуточных продуктов, образующихся на последующих стадиях, повторную валидацию или оценку (при невозможности валидации) при простом изменении допускается, как правило, проводить в отношении измененного этапа. При внесении изменений в производственный процесс, которые затрагивают несколько стадий производственного процесса, может потребоваться более расширенный анализ и, как следствие, валидация.
Подтверждение наличия контроля над измененным процессом производства включает в себя в том числе:
изменение спецификаций на сырье, исходные материалы и реагенты;
надлежащие испытания на биологическую нагрузку и (или) вирусную безопасность измененного банка клеток и клеток с предельным для производства клеточным возрастом in vitro;
очистка от посторонних агентов;
удаление родственных и производственных примесей, например, остаточных ДНК и белков клетки-хозяина;
поддержание необходимого профиля чистоты.
Для зарегистрированных лекарственных препаратов должно быть проанализировано достаточное число серий, произведенных после внесения изменений в процесс производства, с целью подтверждения постоянства производства.
Для обоснования анализа изменений и стратегии контроля качества производитель должен подготовить описание изменения, в котором резюмируется процесс производства до и после изменения и четко обозначены изменения процесса и контроля в параллельном формате.
2.4. Подтверждение сопоставимости на этапе
разработки продукта
При разработке продукта возможно внесение большого числа изменений в производственный процесс, которые могут повлиять на качество лекарственного препарата, его безопасность и эффективность. В целях содействия дальнейшей разработке и регистрации такого продукта проводят сравнительные исследования, подтверждающие, что полученные доклинические и клинические данные о неизмененном лекарственном препарате распространяются на измененный продукт. Исследования сопоставимости продуктов на стадии разработки зависят от таких факторов, как стадия разработки, доступность валидированных аналитических методик, степень изученности продукта и процесса, которые в некоторых случаях ограничены лишь опытом самого производителя.
Если изменения осуществляются до начала доклинических исследований, вопрос о сопоставимости не возникает, поскольку производитель впоследствии проводит доклинические и клинические исследования, используя в процессе разработки измененный лекарственный препарат. На ранних фазах доклинических и клинических исследований изучение сопоставимости, как правило, не столь обширно, как для зарегистрированного лекарственного препарата. По мере накопления знаний и сведений и разработки аналитических инструментов в исследованиях сопоставимости следует использовать всю имеющуюся информацию. Если изменения осуществляются на поздних этапах разработки и дополнительные клинические исследования в обоснование регистрации проводить не планируется, исследования сопоставимости должны быть столь же всесторонними и глубокими, как в случае, если они проводятся в отношении зарегистрированного лекарственного препарата. Некоторые результаты исследований сопоставимости по качеству могут потребовать проведения дополнительных доклинических и клинических исследований (в соответствии с требованиями подразделов 2.1 - 2.3 настоящей главы, а также главы 9.2 настоящих Правил).
При проведении исследований сопоставимости в процессе разработки продукта необходимо использовать соответствующие методы оценки. Следует учитывать, что в процессе разработки аналитические методики могут быть не валидированы, но они должны быть всегда научно обоснованы и должны обеспечивать достоверные и воспроизводимые результаты. Ввиду ограниченности аналитических средств на ранней стадии клинической разработки для установления сопоставимости результатов физико-химических и биологических испытаний может оказаться недостаточно, в связи с чем могут потребоваться связующие доклинические и (или) клинические исследования.
2.5. Доклинические и клинические исследования
2.5.1. Факторы, которые необходимо учитывать при планировании доклинических и клинических исследований.
Если производитель сможет доказать сопоставимость с помощью аналитических исследований, описанных в настоящей главе, возможно подтверждение сопоставимости измененного и неизмененного продукта исключительно с помощью показателей качества (в соответствии с требованиями подраздела 2.2 настоящей главы). Если результатов изучения качества недостаточно, для установления сопоставимости необходимо дополнительно провести доклинические и (или) клинические исследования. Объем и разновидность доклинических и клинических исследований устанавливаются в индивидуальном порядке, принимая во внимание множество факторов, включая в том числе следующее:
результаты изучения качества:
лекарственный препарат - вид, характер и степень различия между продуктом, полученным после внесения изменений, и продуктом до внесения изменений с точки зрения показателей качества, включая родственные соединения, профиль примесей, стабильность и вспомогательные вещества (например, новые примеси могут потребовать проведения токсикологических исследований с целью их квалификации);
результаты оценки (валидации) нового процесса производства, включая результаты значимых внутрипроизводственных испытаний;
доступность, возможности и ограничения испытаний, используемых для оценки сопоставимости;
основные свойства и известные данные о продукте
сложность продукта, включая гетерогенность и структуры более высокого порядка - физико-химические и in vitro биологические испытания не всегда способны обнаруживать все структурные и (или) функциональные различия;
зависимость "структура - активность" и сила связи между показателями качества и безопасностью и эффективностью;
взаимосвязь между терапевтическим белком и эндогенными белками организма и их последствия для иммуногенности;
механизм действия продукта (известен или не известен, один или несколько активных центров);
имеющиеся доклинические и клинические данные, значимые для лекарственного препарата, особенности его применения и фармакотерапевтическая группа:
показания к применению и целевые группы пациентов - выявленные различия могут оказать разное влияние на разные популяции пациентов, например, риск нежелательной иммуногенности. При этом может потребоваться рассмотрение последствий внесенных изменений для каждого показания к применению;
способ применения, например, режим дозирования, путь введения - риск определенных последствий, например, иммуногенности, может быть выше при длительном введении, чем при краткосрочном; подкожное введение чаще предрасполагает к иммуногенности, чем внутривенное;
терапевтический диапазон (кривая "доза - эффект"): влияние определенных изменений на лекарственные препараты с широким терапевтическим диапазоном может отличаться от лекарственных препаратов с узким диапазоном. Даже незначительные изменения фармакокинетики или профиля связывания с рецептором могут оказывать влияние на безопасность и эффективность лекарственных препаратов с крутой или колоколообразной кривой "доза - эффект";
предыдущий опыт в отношении действия лекарственного препарата, например, в отношении его иммуногенности, безопасности. Следует учитывать опыт применения неизмененного лекарственного препарата или лекарственных препаратов того же класса, особенно в отношении редких нежелательных реакций, таких последствия его иммуногенного действия;
фармакокинетическо-фармакодинамическая зависимость, распределение и клиренс.
2.5.2. Тип исследования.
К доклиническим и клиническим исследованиям, указанным в настоящей главе, в зависимости от обстоятельств относятся: ФК-исследования, ФД-исследования, ФК/ФД-исследования, исследования клинической эффективности, исследования клинической безопасности, исследования иммуногенности, исследования в рамках фармаконадзора. Цель указанных исследований - сравнить измененный и неизмененный лекарственный препарат. По возможности такие исследования должны носить прямой сравнительный характер.
3. Определения
Для целей настоящей главы используются понятия (термины), которые означают следующее:
"исследование сопоставимости" - деятельность, включающая в себя планирование исследований, их проведение и анализ данных, направленных на установление сопоставимости лекарственных препаратов;
"показатели качества" - молекулярная характеристика и другие свойства продукта, отобранные в силу их способности определять качество продукта. Показатели качества в совокупности определяют подлинность, чистоту, активность и стабильность продукта, а также безопасность с точки зрения посторонних агентов. Спецификации определяют определенный набор показателей качества;
"связующее исследование сопоставимости" - исследования, обеспечивающие возможность экстраполяции доклинических и клинических данных, полученных при изучении лекарственного продукта, произведенного с помощью неизмененного процесса производства, на лекарственный препарат, произведенный с помощью измененного процесса;
"сопоставимость" - заключение, что продукты обладают высокой степенью сходства по показателям качества продукта до и после изменений процесса производства и отсутствует нежелательное влияние на безопасность или эффективность, включая иммуногенность, лекарственного препарата. Такое заключение можно сделать по результатам анализа показателей качества продукта. В некоторых случаях для такого заключения необходимы доклинические или клинические данные.
Глава 9.2. Исследование сопоставимости биотехнологических
лекарственных препаратов при внесении изменений
в производственный процесс: доклинические
и клинические исследования
1. Введение
Производители биотехнологических (биологических) лекарственных препаратов часто вносят изменения в производственный процесс на этапах разработки и после регистрации.
Подтверждение сопоставимости препаратов, полученных до и после внесения изменений, является последовательным процессом, который начинается с изучения качества (в ограниченном или полном объеме) и, при необходимости, может включать в себя доклинические, клинические исследования и (или) исследования в рамках фармаконадзора.
В настоящей главе представлены требования по проведению доклинических и клинических исследований в рамках исследования сопоставимости при сравнении препаратов, которые были получены до и после изменений процесса производства, внесенных одним производителем, включая контрактных производителей. В настоящей главе представлены требования для проведения связующих доклинических и (или) клинических исследований после внесения изменений в производственный процесс для подтверждения отсутствия влияния таких изменений на эффективность и безопасность препарата.
Программа доклинических и клинических исследований должна основываться на свойствах препарата и быть составлена таким образом, чтобы с достаточной точностью предсказать и выявить возможные различия между сравниваемыми препаратами, которые получены до и после внесения изменения в производственный процесс.
Предполагается, что физико-химические свойства и биологическая активность в условиях in vitro и in vivo препарата могут быть хорошо охарактеризованы с использованием современных методов.
Для большинства изменений, вносимых в производственный процесс, результаты сравнительного изучения физико-химических свойств и биологической активности (как показателя качества) позволяют подтвердить отсутствие различий по показателям качества, которые могут негативно влиять на безопасность и эффективность. Таким образом, исследования сопоставимости ограничиваются только валидацией измененного процесса или расширяются дополнительными критериями качества за счет внутрипроизводственного контроля, физико-химических и биологических характеристик препарата и данных по стабильности (в соответствии с требованиями главы 9.1 настоящих Правил). Однако в некоторых случаях можно ожидать влияние выявленных различий между препаратом до и после изменений на его безопасность и (или) эффективность или наличие таких влияющих различий не может быть исключено, несмотря на современный уровень использованных физико-химических и биологических методов. В подобных ситуациях необходимо провести дополнительные доклинические и (или) клинические исследования.
Вид и объем доклинических и клинических исследований зависят от многих факторов, обусловленных активной фармацевтической субстанцией и лекарственным препаратом, таких как:
информация о молекуле и других молекулах из данной группы препаратов;
этап разработки еще не зарегистрированного препарата;
результаты сравнительных исследований физико-химических и биологических свойств по оценке сопоставимости;
предлагаемое клиническое применение.
2. Область применения
Указанные в настоящей главе принципы распространяются на белки и полипептиды, их производные, а также препараты, в которых они являются компонентами, например, в конъюгатах. Такие белки и полипептиды могут быть получены с помощью рекомбинантных или нерекомбинантных экспрессирующих систем, они хорошо поддаются очистке и подробному установлению характеристик с использованием современных аналитических методов в следующих случаях:
изменения в производственный процесс вносятся одним производителем (в том числе контрактным), который может непосредственно сравнить процессы, результаты аналитических внутрипроизводственных испытаний, полученных до и после внесения изменений в производственный процесс;
изменения в производственный процесс вносятся в процессе разработки или после регистрации лекарственного препарата.
Принципы, указанные в настоящей главе, могут быть применимы к другим биологическим лекарственным препаратам, таким как белки и полипептиды, полученные из тканей и жидкостей организма. В подобных ситуациях производителю необходимо проконсультироваться с уполномоченным органом государства-члена для возможности применения указанных требований.
3. Общие положения
Настоящую главу следует рассматривать в совокупности с другими главами настоящих Правил.
4. Основной текст правил
4.1. Использование подхода, основанного на оценке рисков
для определения потребности в доклинических
и клинических исследованиях
Подтверждение сопоставимости препаратов, полученных до и после внесения изменений в производственный процесс, является последовательным процессом, который начинается с изучения качества (в ограниченном или полном объеме) и при необходимости может включать в себя доклинические, и (или) клинические исследования, и (или) исследования в рамках фармаконадзора. Если производитель может подтвердить сопоставимость с помощью физико-химических и биологических исследований, проведение доклинических и клинических исследований не требуется. В противном случае необходимо провести дополнительные доклинические и (или) клинические исследования.
Необходимость проведения, объем и характер доклинических и клинических исследований сопоставимости определяются в индивидуальном порядке с учетом различных факторов, которые связаны с риском, таких как:
сложность производственного процесса, характер изменения и его потенциальная возможность влияния на структуру молекулы действующего вещества и характеристики лекарственного препарата;
характер и степень различий, обнаруженных с помощью физико-химических и затрагивающих качество биологических испытаний, включая родственные соединения, профиль примесей, стабильность и вспомогательные вещества. Хорошо охарактеризованные различия являются основой для рационального и направленного подхода к определению необходимости для проведения доклинических и клинических исследований;
сложность препарата, включая гетерогенность и структуры более высокого порядка, а также наличие, возможности и ограничения аналитических испытаний. Если аналитические методики не позволяют выявить различия, которые могут повлиять на безопасность и эффективность препарата после внесения изменений в производственный процесс, могут потребоваться дополнительные доклинические и (или) подтверждающие клинические исследования;
структурно-функциональная зависимость и сила связи показателей качества с безопасностью и эффективностью;
взаимосвязь между белковым препаратом и эндогенными белками, и тяжесть потенциальных последствий иммуногенности, например, риск развития аутоиммунных нарушений;
механизмы действия: неизвестные и множественные механизмы действия усложняют оценку влияния изменений;
показания к применению и целевые группы пациентов: различия могут по-разному проявиться в различных группах пациентов при разных показаниях к применению;
способ применения, например, режим дозирования и путь введения (в частности, многократное подкожное введение препарата чаще ассоциировано с иммуногенностью, нежели однократное внутривенное введение);
терапевтический диапазон (кривая "доза - эффект");
предыдущий опыт, например, иммуногенность и безопасность. Актуальным будет анализ данных, полученных до внесения изменений в производственный процесс, или данных о других препаратах из этой группы. Однако биотехнологические белки следует рассматривать индивидуально.
Указанные особенности необходимо учитывать при изучении сопоставимости в процессе разработки препарата. Объем необходимых исследований сопоставимости будет больше при внесении изменений на более поздних этапах клинической разработки. Наиболее сложной проблемой является изучение сопоставимости при внесении изменений в производственный процесс после окончания подтверждающих клинических исследований эффективности и безопасности.
Выбор доклинических и клинических исследований определяется свойствами конкретного препарата, то есть необходимо выбрать такую стратегию проведения исследований сопоставимости, которая оптимальным образом позволит достаточно точно спрогнозировать и выявить все потенциальные клинически значимые различия.
4.2. Доклинические исследования
Если для оценки сопоставимости препаратов до и после изменения результатов одних физико-химических и затрагивающих качество биологических испытаний недостаточно вследствие выявления различий между препаратами или характера изменения процесса производства, не позволяющего по результатам изучения одного лишь качества исключить различия, результаты доклинических исследований могут обнаруживать полезные сигналы потенциальных различий в эффективности и безопасности.
В частных случаях целесообразно провести небольшое число доклинических исследований или не проводить их в принципе, однако в других случаях необходимо провести большой объем исследований. Необходимо отметить, что составление надлежащей программы доклинических исследований требует четкого понимания структуры и активности препарата. При этом необходимо учитывать соответствующие документы, в частности главы 5.3 - 5.4 настоящих Правил. Выявление изменений в профиле примесей необязательно требует проведения доклинических исследований. При этом держатели регистрационных удостоверений должны представить обоснование стратегии (плана) дальнейших действий.
Доклинические исследования сопоставимости носят сравнительный характер, основной целью которых является выявление возможных различий между ответами на препараты, полученные до и после внесения изменений, а не ответа per se. При этом сопоставление препаратов, полученных до и после внесения изменений, необходимо проводить в одном исследовании.
При обосновании подхода к планированию доклинических исследований сопоставимости в модулях 2 и 4 регистрационного досье необходимо представить достаточные сведения и ссылки на другие разделы регистрационного досье. Допускается придерживаться нижеследующего подхода, который следует адаптировать в индивидуальном порядке к рассматриваемому лекарственному препарату. Выбранный подход необходимо обосновать в доклиническом обзоре (включаемом в модуль 2.6. регистрационного досье).
Исследования in vitro
В целях выявления любых произошедших изменений в реактивности и обнаружения вероятных причин несопоставимости необходимо, используя параллельный сравнительный дизайн, изучить препараты до и после внесения изменений с помощью биологических испытаний (например, связывание с рецептором или испытания на клетках), многие из которых могут быть доступны при оценке качества.
Исследования in vivo
При сохранении неопределенности или опасений (обоснованного экспертного мнения или мнения разработчика препарата относительно отсутствия важных данных, сведений, поясняющих полученную информацию и сведения, содержащихся в регистрационном досье, либо наличия в досье взаимно противоречащих сведений и данных) относительно значений (характеристик) фармакокинетических параметров или фармакодинамических эффектов, значимых для клинического применения и (или) безопасности, следует рассмотреть возможность проведения исследований in vivo на одном или нескольких релевантных видах животных с использованием должным образом валидированных животных моделей. Большую достоверность представляют результаты исследований, проведенных на видах животных, в отношении которых на неизмененном препарате показано, что они являются релевантными для человека. При проведении указанных исследований предпочтительно использовать лекарственный препарат, полученный после внесения изменения, нежели активную фармацевтическую субстанцию. В целях упрощения интерпретации данных состав препарата, используемого при проведении доклинических исследований, должен совпадать с составом, который планируется использовать в клинических исследованиях. При проведении доклинических испытаний следует использовать современные методы исследований.
В целом и если позволяет модель, необходимо осуществлять наблюдение за несколькими конечными точками, такими как:
изменения показателей фармакодинамики, значимых для клинического применения (например, длительности действия);
изменения фармакокинетических параметров (например, клиренса);
специально подобранные токсикологические точки (прижизненные и посмертные). Дизайн исследования необходимо обосновать с учетом предполагаемой продолжительности клинического применения;
иммунный ответ, например, титры антител, нейтрализующая активность и перекрестная реактивность. Несмотря на то что прогностическая ценность изучения иммуногенности на животных моделях для человека низкая, в целях упрощения интерпретации результатов исследований токсичности при многократном введении в них необходимо включить (сравнительные) конечные точки иммуногенности и забор образцов крови.
Разработчику лекарственного препарата следует учитывать, что необходимо регулярно пересматривать методы исследования с учетом достижений в области аналитических и биологических методов исследований, например, определенную ценность могут представлять исследования in vitro связывания в режиме реального времени. При проведении исследований in vivo могут быть использованы геномные или протеомные микропанели, которые могут позволить обнаружить незначительные изменения биологического ответа на фармакологически активные вещества.
4.3. Клинические исследования
Программа сравнительных клинических исследований эффективности и безопасности должна быть составлена с учетом этапа разработки препарата, характера вносимых изменений и их влияния на показатели качества. Разработчики как правило вносят изменения в процесс производства препарата до его регистрации. Количество дополнительных данных меньше при внесении изменений до проведения подтверждающих клинических исследований, чем при внесении изменений после проведения подтверждающих клинических исследований или регистрации. При планировании клинических исследований, необходимо учитывать следующие варианты внесения изменений в процесс производства:
изменения процесса производства произошли до начала проведения подтверждающих исследований. В этом случае в целях подтверждения того, что имеющиеся доклинические и клинические данные, полученные до изменения, остаются валидными и возможна их экстраполяция на препарат после изменения, как правило, достаточно представить соответствующие результаты физико-химических и биологических (исследований in vitro и in vivo). В некоторых случаях, необходимы доклинические или клинические исследования сопоставимости, например, такие как фармакокинетические исследования с однократным введением;
изменения процесса производства произошли в ходе подтверждающих исследований. Внесение изменений в ходе подтверждающих исследований не рекомендуется, однако при необходимости внесения таких изменений заявителю следует обратиться за консультацией в уполномоченные органы государств-членов;
изменения процесса производства произошли по окончании подтверждающих исследований или после регистрации. Если изменение производства происходит по окончании подтверждающих исследований или после регистрации, как правило, необходимо провести более глубокие исследования сопоставимости, включая физико-химические и биологические исследования in vitro, если необходимо фармакокинетические и (или) фармакодинамические исследования сопоставимости. Если результаты таких исследований сопоставимости не исключают влияния на профиль эффективности и безопасности препарата, могут потребоваться дополнительные клинические исследования. Отступление от этой концепции необходимо обосновать;
имеются дополнительные критерии, влияющие на необходимость представления сравнительных клинических данных. При подготовке и обосновании программы клинических исследований необходимо учесть все значимые данные, включая все результаты ранее проведенных доклинических исследований и клинический опыт, полученный в отношении неизмененного препарата и других препаратов из той же категории, в том числе:
зависимость эффективности и безопасности от дозы (экспозиции);
наличие динамического маркера, который можно использовать в качестве суррогатного маркера клинической эффективности и безопасности;
зависимость этого суррогатного маркера от дозы (экспозиции);
взаимодействие препарата с рецепторами;
механизм действия, специфичный для заболевания;
органы-мишени для проявления активности и токсичности лекарственного препарата;
способ применения лекарственного препарата.
Фармакокинетические исследования
Фармакокинетические исследования являются важнейшей частью клинического исследования сопоставимости. Поскольку целью исследования сопоставимости является подтверждение сопоставимости препаратов, а не только характеристика клинической фармакологии препарата per se, полученного после внесения изменений в процесс производства, такие исследования должны быть сравнительными.
Наиболее приемлемым является перекрестное исследование при однократном введении препарата, поскольку оно имеет более низкую вариабельность результатов, чем сравнительные исследования с параллельным дизайном. Следует отметить, что при изучении фармакокинетики и фармакодинамики необходимо учитывать такие факторы, как иммуногенность и ее возможное влияние на фармакокинетические параметры и (или) фармакодинамические эффекты.
Путь введения должен совпадать с тем, который планируется использовать при клиническом применении. Если планируется использовать не 1 путь введения препарата (например, внутривенное и подкожное введение), может потребоваться изучение каждого пути введения. В целях выявления значимых различий выбранная доза должна находиться на крутой части кривой "доза - эффект". При выборе популяции для исследований (здоровые добровольцы или пациенты) в первую очередь следует учитывать механизм действия препарата. Поскольку фармакокинетические и фармакодинамические исследования рекомендуется комбинировать, выбор целевой группы должен основываться на том, какие фармакодинамические эффекты подлежат изучению, то есть оптимально ли обнаружение таких эффектов в выбранной целевой группе. При планировании такого перекрестного исследования необходимо учитывать возможность возникновения эффекта переноса.
Дизайн сравнительных ФК-исследований необязательно должен воспроизводить стандартный дизайн клинической сопоставимости (в соответствии с требованиями правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Комиссией), поскольку схожесть абсорбции и биодоступности не единственный интересующий параметр. Необходимо изучить различия между характеристиками элиминации препаратов, например, клиренсом и терминальным периодом полувыведения.
Фармакодинамические исследования
Фармакодинамику (ФД) предпочтительно следует оценивать в составе сравнительного фармакокинетического исследования, поскольку изменение ФД в некоторых случаях может быть обусловлено изменением фармакокинетических показателей. Исследования должны быть сравнительными и не должны быть направлены на выявление фармакодинамических свойств препарата per se.
Маркеры первичной и вторичной ФД. Выбранная конечная точка должна удовлетворять следующим требованиям:
чувствительность, достаточная для выявления даже незначительных различий;
прецизионность, достаточная для выявления даже незначительных различий;
клиническая значимость для исследуемой целевой популяции.
Для подтверждения аналитической чувствительности следует соблюдать осторожность при определении правильного диапазона доз. Целесообразно проводить исследования нескольких доз. Необходимо обосновать выбор маркеров, а также заранее определить и обосновать границу эквивалентности для него.
В связи с этим выбор популяции необходимо обосновать. Выявление определенных первичных или вторичных ФД-маркеров возможно только у пациентов, а не у здоровых добровольцев. Например, иммуномодуляторы, воздействующие на патологически измененные иммунные эффекторные клетки, не всегда будут вызывать аналогичный эффект у здоровых добровольцев.
ФД-маркеры как замена оценки эффективности. При проведении клинических исследований эффективность обычно оценивается по одной или нескольким клиническим конечным точкам. Иногда для этого используются ФД-маркеры. ФД-маркер может рассматриваться как подходящий маркер эффективности, если его изменение под влиянием препарата в значительной степени объясняет изменения клинического исхода.
ФД-маркеры обычно более чувствительны к изменениям активности препарата и поддаются более ранней оценке, чем клинические конечные точки, поэтому в некоторых случаях они могут служить наиболее подходящей конечной точкой. Однако поскольку цель сравнительного исследования заключается в подтверждении эквивалентности препаратов, как правило, необходимо представить данные о количественной зависимости между ФД-маркером и клинической конечной точкой, позволяющей определить и обосновать границу эквивалентности для эффективности. В некоторых ситуациях целесообразно использовать не один, а несколько ФД-маркеров.
Поскольку суррогатный маркер будет полезен для дальнейшей разработки препарата, изучение суррогатных конечных точек заявителям и держателям регистрационных удостоверений следует проводить.
Исследования эффективности
Дизайн исследования. Если подходящие маркеры отсутствуют или с помощью ФД-исследований не удалось однозначно подтвердить сопоставимость, необходимо, используя клинические конечные точки, провести сравнительное клиническое исследование эквивалентности. Исследования должны носить сравнительный характер с сопоставлением препаратов до и после изменений. Для исключения систематических ошибок клинические исследования должны, как правило, быть рандомизированными, двойными слепыми. Потенциальные различия в эффективности необходимо изучить в исследованиях, позволяющих с наибольшей вероятностью обнаружить такие различия.
Приемлемую границу эквивалентности необходимо выбрать заранее, принимая во внимание спецификации препарата на выпуск, клиническую значимость и статистические аспекты. При определении размера выборки необходимо руководствоваться не только соображениями клинической эффективности, но и исходить из необходимости обеспечить обнаружение различий в безопасности (как это указано далее).
Если исследование с дизайном эквивалентности невыполнимо, необходимо рассмотреть возможность использования других дизайнов и проконсультироваться с уполномоченными органами (экспертными организациями).
Выбор наиболее подходящей популяции пациентов и показаний к применению. Поскольку белковые лекарственные препараты могут применяться по нескольким показаниям и (или) у разных популяций пациентов, необходимо учитывать различия и особенности влияния препарата на показатели эффективности и (или) безопасности. Необходимо выбрать такую популяцию пациентов или такое показание к применению, которые наилучшим образом позволят выявить различия, то есть наиболее чувствительную модель оценки эффективности. Выбор популяции заявителем требует обоснования и зависит от ее восприимчивости и подверженности потенциальным рискам для безопасности. Заявитель должен всесторонне с достаточной достоверностью обосновать возможность экстраполяции результатов сравнительного изучения эффективности и безопасности по одному показанию или у одной популяции на другие популяции и показания к применению.
Выбор подходящих конечных точек. Необходимо выбрать такие конечные точки, которые позволяют с высокой степенью точности выявить возможные различия между препаратами. Требования к проведению клинических исследований сопоставимости могут отличаться от требований, которые предъявляются к проведению стандартных подтверждающих исследований. Клинические конечные точки зарегистрированных лекарственных препаратов необязательно должны совпадать с конечными точками, использованными в подтверждающих исследованиях, если их способность обнаруживать различия недостаточна. Более подходящими могут оказаться фармакодинамические или иные маркеры, например, визуализационные. Выбор клинических конечных точек необходимо обосновать.
Продолжительность исследования. Продолжительность исследования определяется главным образом выбором клинической конечной точки. Продолжительность должна быть достаточной, чтобы достаточно точно выявить даже минимальные различия. При обсуждении и обосновании продолжительности исследования необходимо также опираться на данные опубликованные в научной медицинской литературе. Поскольку сравнительная оценка безопасности является неотъемлемой частью исследования клинической сопоставимости, при определении продолжительности исследования следует также учесть необходимость надлежащего выявления значимых различий по показателям безопасности.
Требования к клинической безопасности и фармаконадзору
Если в процессе изучения эффективности доказана сопоставимость препаратов, полученных до и после внесения изменений, то такие препараты могут иметь различия по показателям безопасности (по характеру, серьезности или частоте возникновения нежелательных реакций). Данные по оценке безопасности препарата, не прошедшего регистрацию, необходимо получить в исследовании с участием достаточного числа пациентов, чтобы можно было провести сравнение профилей безопасности препаратов, полученных до и после внесения изменений в процесс производства. Необходимо с осторожностью сравнивать вид, тяжесть и частоту возникновения нежелательных реакций между препаратами до и после изменения.
После регистрации препарата могут потребоваться дополнительные исследования, например, фармакоэпидемиологические.
При оценке нежелательных реакций необходимо учитывать не только частоту возникновения нежелательных явлений, но и возможные различия в их клинических проявлениях (длительность, тяжесть и серьезность, обратимость, ответ на лечение и т.д.).
Заявитель должен включить в регистрационное досье рассматриваемого лекарственного препарата спецификацию по безопасности. Она должна содержать описание возможных проблем с безопасностью, обусловленных изменениями процесса производства.
Объем базы данных по оценке безопасности. Данные о безопасности могут быть получены в рамках клинического исследования, направленного на подтверждение эквивалентной эффективности. Продолжительность исследования и объем выборки определяют с учетом частоты возникновения, тяжести и серьезности ожидаемых нежелательных явлений, а также клинических условий применения лекарственного препарата (непродолжительное или длительное применение и др.). Цель такого исследования не заключается в выявлении нежелательных явлений per se, она состоит в оценке различий в их возникновении.
Конечные точки безопасности. При выборе конкретных конечных точек следует учитывать как типичные, характерные для препарата и (или) его класса аспекты безопасности, так и прочие потенциальные проблемы, которые можно вывести исходя из механизма действия. Поскольку можно получить неожиданные результаты изучения безопасности, заявителям не рекомендуется в протоколе исследования ограничиваться методами, направленными на обнаружение исключительно известных проблем для безопасности. Оценка сравнительной иммуногенности должна быть составной частью оценки безопасности (в соответствии с требованиями главы 11 настоящих Правил).
План управления рисками
При регистрации заявитель должен представить план управления рисками или после регистрации препарата представить обновленный план управления рисками в соответствии с международными договорами и актами, составляющими право Союза. При этом необходимо учесть риски, которые были выявлены в процессе изучения безопасности препарата, и потенциальные риски.
При разработке плана управления рисками следует учитывать уже существующую информацию по безопасности применения ранее выпускавшегося препарата (препарата, полученного по исходной технологии до внесения изменений в процесс производства), а также лекарственных препаратов данной группы.
В периодических отчетах по безопасности лекарственных препаратов (ПООБ), регулярно представляемых держателем регистрационного удостоверения согласно праву Союза, должна содержаться вся информация о рисках и переносимости препарата, зависящих от внесенных изменений в процесс производства. Периодичность представления ПООБ определяется в каждом конкретном случае индивидуально.
Сроки представления доклинических и (или) клинических данных. Доклинические и клинические исследования (если применимо) должны быть завершены до утверждения изменений производственного процесса в соответствии с установленными в Союзе требованиями, то есть до выпуска препарата. В зависимости от свойств препарата и показаний к применению вносимые изменения могут быть основаны на фармакодинамических данных. При этом дополнительные данные клинических исследований и данные по безопасности, включая сведения об иммуногенности, могут быть представлены после утверждения вносимых изменений.
Глава 10. Разработка, производство, установление
характеристик и спецификации моноклональных антител
и их производных
1. Введение
В настоящей главе рассматриваются требования к качеству моноклональных антител.
Моноклональные антитела - это Ig, характеризующиеся определенной специфичностью, источником получения которых являются линии клеток одного клона. Их биологическая активность проявляется за счет специфичного связывания с соответствующим лигандом (обычно определяемым как антиген) и обусловливает такие эффекторные функции иммунной системы, как антителозависимая клеточная цитотоксичность (ADCC) и комплементзависимая цитотоксичность (CDC).
Моноклональные антитела могут быть получены по технологии рекомбинантной ДНК (рДНК), гибридомной технологии, иммортализацией B-лимфоцитов или с помощью других технологий (например, дисплей-технологии, генетически модифицированные животные).
В настоящей главе изложены принципы и общие требования к разработке, производству, установлению характеристик и спецификации препаратов моноклональных антител, которые применяются в качестве лекарственных препаратов для медицинского применения или использования в их производстве.
2. Область применения
В настоящей главе рассматриваются вопросы качества при регистрации моноклональных антител, полученных из моноклональной линии клеток, предназначенных для терапевтического и профилактического (в том числе для применения ex vivo), а также для диагностического применения in vivo.
Принципы, изложенные в настоящей главе, применимы к моноклональным антителам, используемым в качестве реагентов, а также к лекарственным препаратам, разработанным на основе моноклональных антител, таким как фрагменты иммуноглобулинов, конъюгаты, гибридные белки и др. Однако использование указанных принципов определяется индивидуально для каждого конкретного препарата с учетом специфики их свойств и будет рассмотрено в отдельных документах.
Поликлональные антитела (фракционированные и рекомбинантные) в настоящей главе не рассматриваются, однако по возможности следует использовать описанные в них принципы.
Настоящая глава не распространяется на:
моноклональные антитела, предназначенные для использования in vitro;
моноклональные антитела, применяемые в клинических исследованиях.
Однако при производстве и контроле моноклональных антител для клинических исследований необходимо учитывать принципы, описанные в настоящей главе; их применимость будет определяться в индивидуальном порядке.
3. Общие положения
Настоящая глава неразрывно связана с другими главами настоящих Правил, а также с требованиями Фармакопеи Союза ("Моноклональные антитела для клинического применения").
4. Основные положения
4.1. Разработка моноклональных антител
Структуру моноклонального антитела необходимо обосновать с учетом механизма действия, биологической активности и стабильности препарата. Обоснование характеристики структуры моноклональных антител должно содержать по крайней мере рассмотрение пригодности иммунохимических свойств иммуноглобулинов (таких как аффинность, перекрестная реактивность, изотип, аллотип), а также важности и сохранности эффекторной функции. Кроме того, необходимо тщательно рассмотреть риск индукции иммунного ответа у пациентов, особенно если препарат не обладает высокой гомологией с иммуноглобулином человека или при выявлении в структуре потенциально иммуногенных эпитопов, поскольку это может привести к клиническим нежелательным реакциям и (или) изменению терапевтического потенциала.
Клеточный субстрат, используемый для получения моноклональных антител, должен представлять собой стабильную, непрерывно культивируемую линию клеток одного клона, разработанную по технологии рекомбинантной ДНК и (или) другим соответствующим технологиям. Основанием для выбора клеточного субстрата является оценка возможности получения продукта желаемого качества по сравнению с возможностью использования других соответствующих подходов.
Если в качестве субстрата используются клетки, полученные по технологии рекомбинантной ДНК, и характеристика системы, используемой для производства антител, должна соответствовать принципам, указанным в главах 1, 2, 5.1 и 5.2 настоящих Правил.
Если до получения моноклональной клеточной линии в ходе разработки осуществляется одна или более специфичных процедур, например, гибридизация клеток, вирусная трансформация, генная библиотека скрининга в фаговом дисплее, использование технологий in silico, in vitro или in vivo, такие методики не требуют подробного описания. Однако необходимо представить достаточный объем сведений об этих процедурах, позволяющий оценить подлинность и чистоту моноклональной клеточной линии, значимых для безопасности и эффективности препарата (например, аминокислотные или посттрансляционные модификации, направленные на модуляцию иммуногенности или эффекторных функций, и сведения о посторонних агентах и потенциальных контаминантах).
Для получения стабильной и непрерывно культивируемой линии клеток, которая будет использована для производства антител может потребоваться иммортализация B-лимфоцитов человека или клеток другого происхождения путем слияния или трансформации клеток. Необходимо тщательно проанализировать выбранный подход с позиций безопасности и эффективности и должным образом обосновать его.
Использование B-лимфоцитов человека в качестве родительских клеточных линий поднимает проблемы, связанные с возможной передачей инфекционных агентов, в том числе агентов вариантной болезни Крейтцфельдта-Якоба, а также других патогенных для человека микроорганизмов. Использование лимфоцитов человека, трансформированных вирусом Эпштейна-Барр (EBV), создает дополнительные трудности в связи с наличием вируса EBV, способного инфицировать человека.
Гибридома, полученная путем гибридизации B-лимфоцитов человека или клеток другого происхождения с миеломными клетками, может быть использована в качестве клеточного субстрата. Происхождение и установление характеристик родительских клеток необходимо подробно описать и документировать, включая информацию о здоровье доноров, использованных партнерах гибридизации и материалах человеческого или животного происхождения, которые соприкасались с клетками (например, питающие клетки и миеломные клетки).
4.2. Производство моноклональных антител
4.2.1. Общие положения.
Процесс производства необходимо должным образом описать и валидировать. Валидация должна по меньшей мере включать в себя:
подтверждение того, что процесс способен производить продукт постоянного качества в соответствии с надлежащим образом заданной стратегией контроля качества;
оценку производственных возможностей (например, элиминацию производственных примесей, вирусов);
подтверждение того, что каждая операционная единица функционирует должным образом (например, валидация очистки колонок, асептическая фасовка).
Внимание должно быть направлено на обеспечение внутрипроизводственного контроля (включая показатели качества промежуточных продуктов и параметры процесса), а также на составление спецификаций на активную фармацевтическую субстанцию и готовый лекарственный препарат. Подобный контроль должен позволять отслеживать значимые показатели качества, такие как родственные соединения и примеси (например, правильность или неправильность дисульфидных связей, дезаминирование, окисление, укорочение, агрегаты) или производственные примеси (например, белки, ДНК, белок A клетки-хозяина, бычья сыворотка, остатки питательных сред), а также релевантные параметры процесса (например, загрузка колонки, pH, температура).
Если белок A используется в процессе очистки, источник белка A (например, S. aureus или рекомбинантный белок) и способ его получения (например, очищенный с использованием IgG человека) должны быть надлежащим образом документированы. Если в производстве использовался IgG человека, необходимо подтвердить, что качество IgG пригодно для целевого назначения, особенно с позиций вирусной безопасности.
4.2.2. Платформенное производство
Разработка процессов, используемых для производства моноклональных антител, во многом зависит от знания производителем как продукта, так и процесса производства.
Некоторые производители приобрели значительный опыт в области производства моноклональных антител и разработали стратегию производства, основанную на схожих производственных процессах (то есть с использованием определенных клеток-хозяина, культуры клеток, процессов очистки целевого белка и т.д.). Такой подход часто называют платформенным производством.
Подобно любому другому лекарственному препарату, процесс производства биотехнологического лекарственного препарата, который был разработан с использованием платформенного производства, должен быть валидирован к моменту его регистрации. Исследования по валидации должны включать в себя данные, полученные от конечного процесса производства и производственных площадок, которые будут использоваться для производства лекарственного препарата для реализации. Однако при должном обосновании и документировании в целях обоснования или снижения подаваемого объема данных, полученных по результатам конечного коммерческого процесса, допускается представить в уполномоченные органы государств-членов результаты, полученные на основании иного релевантного опыта.
С учетом того, что показатели качества специфичны для каждого препарата и процесса его производства, необходимо в отношении регистрируемого препарата и процесса отдельно подтвердить пригодность аналитических методов и стратегии контроля качества в целом. Как следствие, необходимо тщательно пересмотреть пригодность стратегии контроля качества, являющейся пригодной для анализа других препаратов, полученных с помощью того же подхода платформенного производства, поскольку она может быть не адаптирована к регистрируемым препарату и процессу. Например, производственные примеси, такие как белки клетки-хозяина (БКХ), высоко зависимы от процесса, и методы контроля, использующиеся в отношении данного препарата и процесса, могут оказаться непригодными для других продуктов, использующих то же платформенное производство (например, различные клеточные субстраты, полученные из общей парентеральной клеточной линии, аналогичные культуры и условия очистки).
При изменении утвержденного процесса, основанного на платформенном производстве, необходимо отдельно проанализировать влияние такого изменения на рассматриваемые препарат и процесс. Тем не менее при должном обосновании и документировании в целях обоснования или снижения подаваемого объема данных, полученных в отношении измененных препарата и процесса, допускается представить результаты, полученные на основании иного релевантного опыта. Более того, если с помощью общего платформенного процесса производства получают несколько препаратов, а изменения (например, оптимизация или улучшение процесса) вводятся лишь в один или несколько из них, необходимо представить обоснование принятой стратегии гармонизации или отсутствия таковой.
4.2.3. Вирусная безопасность и трансмиссивная губчатая энцефалопатия.
Вопросы вирусной безопасности моноклональных антител, рассматриваемых в настоящей главе, должны соответствовать положениям главы 2 настоящих Правил. Требования, указанные в настоящей главе, касаются моноклональных антител, полученных из гибридомных клеточных линий или генетически модифицированных клеток, продуцирующих моноклональные антитела. Если производство моноклональных антител проводится с использованием животных (например, трансгенных животных или асцитической жидкости), необходимо учитывать требования главы 2 настоящих Правил, в частности приложения N 1 к указанной главе. Исходные клетки (например, клетки-хозяина) должны пройти скрининг на посторонние агенты, то есть на наличие посторонних или эндогенных агентов. Выбор вирусов, которые следует использовать в испытаниях, зависит от вида животных и ткани-источника клеток-продуцентов, а также свойств любого другого биологического сырья, используемого в производстве.
Необходимо в обязательном порядке провести надлежащие валидационные исследования по снижению вирусной нагрузки. В соответствии с главой 2 настоящих Правил, в отношении заявленного на регистрацию лекарственного препарата и его процесса производства необходимо валидировать способность производственных стадий снижать вирусную нагрузку. В целях учета потенциальных и неожиданных препаратспецифичных факторов, влияющих на снижение вирусной нагрузки, подобные валидационные исследования, как правило, проводят с использованием промежуточных продуктов, получаемых в отдельном процессе производства. Тем не менее при должном обосновании и документировании в целях установления и анализа стадий по снижению вирусной нагрузки ценными являются и иные исследования (например, проведенные на основании подхода платформенного производства), которые могут позволить снизить объем подаваемых результатов валидационных исследований. Такие данные можно рассматривать как вспомогательные (например, при изучении потенциального влияния изменяющихся параметров процесса на снижение вирусной нагрузки, свойств колонок после множества производственных циклов, исследований переноса вирусов или исследований по очистке колонок). Во всех случаях производитель должен обосновать значимость таких данных для отдельного продукта. Необходимо представить обоснования возможности использования предварительных собственных данных в отношении нового препарата (например, допустимы ли ссылки на данные по снижению вирусной нагрузки на определенных стадиях процесса, если промежуточный продукт, получаемый на предыдущем этапе, обладает сопоставимыми биохимическими свойствами и подвергается очистке идентичными методами). Производитель должен представить критический анализ производственной стадии, в отношении которой будут использоваться подобные собственные вспомогательные данные, и состава соответствующего промежуточного продукта. По результатам анализа необходимо прийти к однозначному заключению, что в обоих случаях введенная производственная стадия аналогична по способности инактивировать (элиминировать) потенциальные вирусные контаминанты. Если сопоставимость этапов неубедительна или база данных не позволяет исключить препаратспецифичное влияние на способность снижать вирусную нагрузку, необходимо провести подтверждающие циклы, используя препарат-специфичные промежуточные продукты.
Если в разработке или производстве использовались материалы крупного рогатого скота или иных видов животных, источников ТГЭ, необходимо следовать требованиям актов, входящих в право Союза, по минимизации риска передачи агентов губчатой энцефалопатии животных посредством лекарственных препаратов для медицинского применения.
4.3. Установление характеристик моноклональные антитела
Моноклональные антитела необходимо подробно охарактеризовать. В соответствии с главой 6 настоящих Правил, установление характеристик должно включать в себя определение физико-химических и иммунохимических свойств, биологической активности, чистоты, примесей и количественного содержания моноклональных антител. На момент регистрации лекарственного препарата заявитель должен располагать соответствующим образом охарактеризованными собственными стандартными материалами, которые будут использоваться в биологических и физико-химических испытаниях промышленных серий.
4.3.1. Физико-химические характеристики.
Программа установления физико-химических характеристик, как правило, включает в себя определение класса, подкласса, строение легких цепей (каппа и (или) лямбда цепи) и первичной структуры моноклонального антитела.
По результатам секвенирования ДНК необходимо вывести аминокислотную последовательность и с помощью надлежащих методов (например, пептидного картирования, секвенирования аминокислот, масс-спектрометрического анализа) подтвердить ее экспериментально. Необходимо проанализировать вариацию N- и C-концевых последовательностей аминокислот (например, C-концевых лизинов).
Необходимо определить свободные сульфгидрильные группы и дисульфидные мостики, сохранность или правильность дисульфидных связей.
Необходимо установить содержание углеводов (нейтральные сахара, аминосахара и сиаловые кислоты). Кроме того, необходимо проанализировать структуру углеводных цепей, олигосахаридный профиль (профиль ветвления), участки гликозилирования и их занятость.
Как правило, моноклональные антитела имеют один участок N-гликозилирования на каждой тяжелой цепи, расположенный в Fc-фрагменте. Легкая цепь, как правило, не гликозилируется. Однако тяжелые цепи могут содержать дополнительные участки гликозилирования, поэтому необходимо установить их наличие или отсутствие. Необходимо описать структуру гликанов, уделив особое внимание степени маннозилирования, галактозилирования, фукозилирования и сиалилирования. Необходимо определить распределение основных имеющихся гликановых структур (чаще всего G0, G1 и G2).
С помощью подходящей методологии необходимо описать структуру моноклонального антитела высшего порядка.
4.3.2. Иммунологические свойства.
Иммунологические свойства антител необходимо всесторонне охарактеризовать. В целях определения их аффинности, авидности и иммунореактивности (включая перекрестную реактивность с другими структурно гомологичными белками) необходимо провести анализ связывания антител с очищенными антигенами и определенными участками антигенов. Необходимо изучить непредусмотренную реактивность (цитотоксичность) для тканей человека, отличных от выбранных мишеней. Используя иммуногистохимические методики в соответствии с приложением к настоящей главе, необходимо установить перекрестную реактивность с тканями человека. В соответствующих случаях допускаются перекрестные ссылки на доклинический и (или) клинический разделы регистрационного досье.
При отсутствии должного обоснования необходимо идентифицировать области, определяющие комплементарность (гипервариабельные участки, CDR).
Необходимо определить эпитоп и молекулу, его несущую. Указанное должно включать в себя биохимическую идентификацию таких структур (например, белок, олигосахарид, гликопротеин, гликолипид) и все возможные исследования по установлению характеристик (аминокислотная последовательность, структура углеводов).
Необходимо изучить способность связываться с комплементом и активировать его и (или) иные эффекторные функции, даже если целевая биологическая активность не требует наличия таких функций.
4.3.3. Биологическая активность.
С помощью исследований in vitro и (или) in vivo необходимо определить биологическую активность (то есть специфическую способность препарата оказывать определенный биологический эффект). Необходимо проанализировать механизм действия и значение (последствия) эффекторных функций препарата для его безопасности и эффективности.
Если эффекторная функция антител может являться частью механизма действия и (или) влиять на безопасность и эффективность препарата, необходимо представить подробный анализ АЗКЦ, цитотоксических свойств (например, апоптоза), способности связываться с комплементом и активировать его, прочие эффекторные функции, включая активность связывания с гамма Fc-рецепторами и неонатальными Fc-рецепторами.
4.3.4. Чистота, примеси и контаминаты.
Как правило, выделяют несколько источников гетерогенности моноклональных антител (например, изменение C-концевого лизина, образование N-концевого пироглутамата, дезамидирование, окисление, изомеризация, фрагментация, образование нетипичных дисульфидных связей, N-связанный олигосахарид, гликирование), которые приводят к сложному профилю показателей чистоты (примесей), представляющему собой несколько молекулярных структур и вариантов. Такой профиль чистоты (примесей) необходимо оценить с помощью совокупности ортогональных методов и для соответствующих родственных вариантов предусмотреть индивидуальные или суммарные критерии приемлемости.
К таким методам, как правило, относятся определение физико-химических свойств, например, молекулярной массы или размера, профиля изоформ, коэффициента экстинкции, электрофоретических профилей, хроматографических данных и спектроскопических профилей. Кроме того, необходимо предложить подходящие методы для качественного и количественного анализа гетерогенности заряженных вариантов.
Используя комбинацию методов, необходимо должным образом охарактеризовать мультимеры и агрегаты. Образование в лекарственном препарате агрегатов, видимых и невидимых включений является важным и требует изучения и тщательного контроля при выпуске серий и в ходе исследований стабильности. В дополнение к фармакопейным испытаниям на механические включения в целях установления природы включений и их содержания могут потребоваться иные ортогональные аналитические методы.
Необходимо идентифицировать потенциальные производственные примеси (например, БКХ, ДНК клетки-хозяина, остаточное содержание питательных сред, остаточное содержание реактивов, использующихся на последующих этапах обработки) и в зависимости от обстоятельств проанализировать их качественно и (или) количественно.
Необходимо строго избегать и (или) должным образом контролировать содержание контаминантов, включающих в себя все привнесенные посторонние материалы, не являющиеся частью процесса производства (например, микроорганизмы, эндотоксины). При подозрении на наличие провоспалительных контаминантов неэндотоксиновой природы (например, пептидогликанов) необходимо провести дополнительные испытания (например, испытание на активацию моноцитов).
4.3.5. Количественное содержание.
С помощью соответствующих физико-химических и (или) иммунохимических методик необходимо определить количественное содержание.
Необходимо подтвердить, что результаты испытания на содержание непосредственно коррелируют с результатами, полученными при испытании на биологическую активность. При наличии такой зависимости в информации о препарате или производственных процессах (например, фасовке) вместо меры биологической активности допускается использовать меру количественного содержания.
4.4. Спецификации
Спецификации являются составной частью общей стратегии контроля качества продукта, которые составляются для обеспечения его качества и постоянства, при этом продукт должен соответствовать спецификации. Необходимо разработать такие спецификации, которые учитывали бы показатели качества, оказавшиеся значимыми по результатам исследований по установлению характеристик. Выбор включаемых в спецификацию испытаний носит препаратспецифичный характер. Необходимо описать основания установления допустимых диапазонов для критериев приемлемости. В соответствии с главой 6 настоящих Правил необходимо установить критерии приемлемости и обосновать их, учитывая данные, полученные по результатам испытаний серий, изученных в доклинических и (или) клинических исследованиях, серий, использованных при подтверждении воспроизводимости процесса производства, данных результатов исследований стабильности и значимых данных по разработке.
4.4.1. Подлинность.
Испытания на подлинность должны быть высоко специфичными и основываться на уникальности молекулярной структуры препарата и (или) иных специфичных свойствах (например, пептидное картирование, антиидиотипный иммуноанализ или иной подходящий метод). С учетом высокой аналогичности константных доменов различных антител в целях установления подлинности могут потребоваться несколько испытаний (физико-химических, биологических и (или) иммунохимических); такие испытания должны позволять различать прочие антитела, которые могут производиться на той же производственной площадке.
4.4.2. Чистота и примеси.
В соответствии с разделом 4.3 настоящей главы профиль чистоты (примесей) моноклональных антител может быть сложным и требует анализа с помощью совокупности ортогональных методов, для которых необходимо установить индивидуальные и (или) суммарные критерии приемлемости по родственным вариантам. Например, в целях качественного и количественного выявления заряженных вариантов необходимо использовать методы разделения, основанные на гетерогенности заряда.
Необходимо включить хроматографические и (или) электрофоретические методы, способные обнаружить укорочение, диссоциацию и полимеризацию продукта, а также предложить для указанных показателей количественные пределы.
Необходимо уделить особое внимание подтверждению пригодности использованных аналитических методик для контроля содержания мультимеров и агрегатов.
Принимая во внимание, что гликозилирование может оказывать влияние на фармакокинетику препарата и изменять его иммуногенные свойства, для гликозилирования необходимо установить надлежащие критерии приемлемости. Кроме того, такой контроль в дальнейшем позволит подтверждать постоянство серий препарата.
Как следствие, необходимо тщательно подобрать испытания и критерии приемлемости гликозилирования (например, относительные количества G0, G1 и (или) G2 Fc-фрагментов, степеней гликозилирования, фукозилирования и сиалилирования), принимая во внимание предусмотренное и потенциальное влияния этого показателя на биологическую активность в клинической практике (например, наличие функциональных эффекторных функций, ненужных для реализации целевого механизма действия, гликозилирование Fab-фрагмента).
В стратегию контроля необходимо включить контроль значимых производственных примесей. В некоторых случаях, при должном подтверждении, контроль их содержания допускается осуществлять на промежуточном продукте, на соответствующем этапе производственного процесса. Для некоторых примесей, для которых показано, что с помощью процесса производства удается значительно сократить их содержание, стандартные испытания не требуются. Контроль остаточного белка A, БКХ, остаточной ДНК и иных потенциальных остаточных содержаний сред или очистки, как правило, является частью спецификации. Кроме того, такой контроль служит ценным источником информации по постоянству и функционированию процесса производства.
4.4.3. Активность.
Активность (potency) - количественная мера биологической активности, основанная на показателе качества препарата, связанном с его значимыми биологическими свойствами. Соответствующая аналитическая методика определения активности должна являться частью спецификаций на активную фармацевтическую субстанцию и (или) лекарственный препарат и в идеальном случае должна отражать биологическую активность в клинической практике.
В отношении антител, клиническая активность которых зависит исключительно от связывающих (нейтрализующих) свойств, при достаточном обосновании допускается использовать аналитическую методику определения активности, определяющую связывание с мишенью (то есть методика связывания). Если для клинической активности необходимы эффекторные функции, необходимо использовать биологический метод количественного определения на основе клеток или иную методику, позволяющую определить эффекторные функции. Если биологический метод количественного определения на основе клеток неуместен или комбинация двух методов дает более точные результаты, следует использовать комбинацию двух отдельных методов: первый - с целью определения специфичности, второй - для определения эффекторной функции (например, активация комплемента, связывание с C1q, связывание с Fc-гамма-рецептором).
Несмотря на то что два вида методик определения активности (связывания и на основе клеток) зачастую дают сопоставимые результаты, такие методики не следует считать взаимозаменяемыми, поскольку некоторые свойства препарата могут не влиять на связывание с мишенью (например, гликозилирование, фрагментация), но влиять на дальнейшее распространение сигнала или экспрессию рецептора.
В целях подтверждения постоянства процесса производства значительную ценность представляет специфическая активность (биологическая активность на массу).
4.4.4. Количественное содержание.
Используя подходящую методику, необходимо определить содержание фармацевтической субстанции, как правило, по содержанию (массе) белка.
4.4.5. Общие показатели.
При необходимости следует изучить внешний вид, растворимость, pH, осмоляльность, извлекаемый объем, стерильность, бактериальные эндотоксины, стабилизатор и воду.
Содержание видимых и невидимых механических включений в лекарственном препарате должно соответствовать требованиям, предъявляемым Фармакопеей Союза.
5. Препараты на основе модифицированных
моноклональных антител
Помимо интактных, немодифицированных моноклональных антител принципы, указанные в настоящих Правилах, могут быть применимы к иным производным от моноклональных антител препаратам (например, фрагменты антител (включая одноцепочечные вариабельные фрагменты (scFv)), гибридные белки, конъюгированные моноклональные антитела, биспецифичные антитела и радиоактивно меченые антитела). Однако их применимость будет определяться в индивидуальном порядке на основании свойств отдельного препарата.
Приложение
к главе 10 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ПЕРЕЧЕНЬ
ТКАНЕЙ ЧЕЛОВЕКА, РЕКОМЕНДУЕМЫХ ДЛЯ ИСПОЛЬЗОВАНИЯ
В ИММУНОГИСТОХИМИЧЕСКИХ ИЛИ ЦИТОХИМИЧЕСКИХ ИССЛЕДОВАНИЯХ
ПЕРЕКРЕСТНОЙ РЕАКТИВНОСТИ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ
Настоящий перечень тканей человека позволяет отразить специфичность антител в иммуногистохимических или цитохимических исследованиях перекрестной реактивности и их клиническое практическое значение и включает в себя в том числе:
миндалину, тимус, лимфатический узел;
костный мозг, клетки крови;
легкие, печень, почки, мочевой пузырь, селезенку, желудок, включая подлежащие гладкие мышцы, кишечник;
поджелудочную железу, большую слюнную железу, щитовидную железу, паращитовидную железу, надпочечник, гипофиз;
головной мозг, периферический нерв;
сердце, поперечнополосатую мышцу;
яичник, яичко;
кожу;
кровеносные сосуды.
Глава 11. Оценка иммуногенности терапевтических белков,
полученных с использованием биотехнологических методов
Количество лекарственных препаратов, являющихся биологическими белками (полученными биотехнологическим путем), неуклонно растет. Они могут вызывать у пациентов нежелательный иммунный ответ, на который влияют различные факторы, в том числе зависящие от пациента, опосредованные заболеванием или лекарственным препаратом. Настоящие Правила содержит указания о выявлении потенциальных причин возникновения и устранении последствий иммуногенности, а также общие указания по проведению систематической оценки иммуногенности при регистрации лекарственных препаратов.
Ввиду неизбежного возникновения у животных иммунного ответа к белкам человека прогностическая ценность доклинических исследований для оценки иммуногенности биологического лекарственного препарата у человека низкая. Несмотря на то что проведение доклинических исследований, направленных на прогнозирование иммуногенности у человека, как правило, не требуется, животные модели могут представлять ценность для оценки последствий иммунного ответа.
В целях измерения иммунного ответа на терапевтические белки необходимо выбрать надлежащую стратегию разработки подходящих методов скрининга и подтверждения наличия антител. Методы должны позволять отличать нейтрализующие антитела от антител, не обладающих такими свойствами, их использование как в основных (опорных) клинических исследованиях, так и в рамках пострегистрационного наблюдения должно быть валидировано.
При тщательном планировании оценки иммуногенности в клиническом центре необходимо систематически набирать данные от достаточного числа пациентов. Предпочтительно, чтобы отбор проб препарата был стандартизирован на протяжении всего исследования (например, отбор проб до начала, во время и по завершении исследования). Схему отбора проб каждого препарата необходимо определять в индивидуальном порядке, учитывая также риски, обусловленные нежелательным иммунным ответом у пациентов. В целях полного понимания клинических последствий иммунного ответа необходимо получить данные о его влиянии на эффективность и безопасность. В последующем вопросы предупреждения иммуногенности необходимо осветить в плане управления рисками.
Требования настоящей главы применимы для различных видов лекарственных препаратов, поэтому общие концепции изучения иммуногенности необходимо адаптировать к каждой отдельной программе разработки в индивидуальном порядке. За разъяснениями заявителям необходимо обращаться за консультацией в уполномоченный орган государства-члена.
1. Введение
Большинство биологических белков (полученных биотехнологическим путем) вызывают нежелательный иммунный ответ, который обусловлен рядом факторов. Иммунный ответ - сложное явление, которое помимо образования антител проявляется активацией T-клеток и врожденного иммунитета, влияющих на развитие нежелательных реакций.
Последствия иммунного ответа на терапевтические белки проявляются изменениями от преходящего появления антител без клинических проявлений и вплоть до тяжелых угрожающих жизни состояний. Потенциальными клиническими последствиями нежелательного иммунного ответа являются снижение эффективности терапевтических белков, тяжелые общие иммунные реакции, включая анафилаксию, и для терапевтических белков, применяемых в качестве заместительной терапии, - потенциальная перекрестная реактивность с эндогенным аналогом, если продукция этого аналога сохранилась.
На иммуногенность терапевтических белков влияет множество факторов. Их можно разделить на зависящие от пациента и опосредованные заболеванием или лекарственным препаратом. Зависящие от пациента факторы могут предрасполагать к развитию иммунного ответа у субъекта и включают в себя: основное заболевание, наследственную предрасположенность, иммунный статус, включая иммуномодулирующую терапию и режим дозирования. Опосредованные лекарственным препаратом факторы также могут влиять на вероятность развития иммунного ответа, например, процесс производства, лекарственная форма и состав лекарственного препарата, его стабильность.
Несмотря на то что данные о возможном нежелательном иммунном ответе на терапевтические белки необходимо представить до регистрации, проблемы с иммуногенностью препарата могут возникнуть и на пострегистрационном этапе. В регистрационном досье необходимо представить резюме об изучении иммуногенности в соответствующих обзорных разделах со ссылками на полные данные в соответствующих модулях. В зависимости от иммуногенного потенциала терапевтического белка и редкости заболевания предрегистрационные данные об иммуногенности могут быть ограничены. После регистрации может потребоваться дальнейшая систематическая оценка иммуногенности, которую можно предусмотреть в плане управления рисками.
2. Область применения
В настоящей главе вводятся общие требования, которые преимущественно касаются установления факта развития нежелательного иммунного ответа на терапевтические белки у пациентов и способов систематической его оценки. Настоящая глава распространяется на белки и полипептиды, их производные, а также препараты, в которых указанные вещества являются компонентами, например, конъюгаты. Эти белки и полипептиды главным образом получают на рекомбинантных или нерекомбинантных экспрессирующих системах. В настоящей главе для их обозначения используется термин "терапевтический белок".
Факторы свертывания крови в настоящей главе не рассматриваются.
3. Общие положения
Настоящая глава неразрывно связана с другими главами настоящих Правил, в частности с главами 9.1, 9.2 и 15 настоящих Правил.
4. Основные положения
Последствия развития иммунного ответа на терапевтические белки могут быть различными - от преходящего выявления антител, не сопровождающегося какими-либо значимыми клиническими явлениями, до тяжелых, угрожающих жизни состояний. Как правило, терапевтические белки следует рассматривать как отдельные лекарственные препараты, опыт применения родственных белков может рассматриваться только в качестве вспомогательных данных. При этом следует учитывать сопутствующую терапию и другие зависящие от пациента факторы, в том числе основное заболевание, поскольку все это также может влиять на клинические проявления иммуногенности. Поэтому оценку иммуногенности необходимо осуществлять в индивидуальном порядке по каждому показанию к применению и популяции пациентов.
Оценка иммуногенности требует междисциплинарного подхода, объединяющего совместные усилия специалистов по обеспечению качества, доклинической и клинической разработке.
В настоящей главе представлены рекомендации и принципы для разработчиков терапевтических белков, получаемых с использованием биотехнологических методов, в целях регистрации экспертов. Указанные положения имеют широкую сферу применения, поэтому с целью создания оптимальной программы разработки допускается адаптировать указанные ниже принципы применительно к конкретному лекарственному препарату. При обосновании выбранного подхода к исследованию иммуногенности заявители должны учитывать как изучение риска развития нежелательного иммунного ответа, так и возможные клинические последствия, указанные ниже. Необходимо полностью обосновать подход к плану изучения иммуногенности, в том числе при отказе от проведения определенных методик или замеров, рекомендуемых в соответствии с настоящей главой. При необходимости заявители вправе обратиться за научной консультацией в уполномоченные органы государств-членов.
4.1. Факторы, влияющие на развитие иммунного ответа
против терапевтических белков
4.1.1. Факторы, зависящие от пациента или опосредованные заболеванием.
К зависящим от пациента факторам, которые могут влиять на иммунный ответ к терапевтическим белкам, относятся генетические особенности, возраст пациента, к опосредованным заболеванием факторам - сопутствующая терапия и введение аналогичных белков ранее.
Генетические факторы, модулирующие иммунный ответ. Генетические факторы могут изменить иммунный ответ на терапевтический белок и могут быть причиной межиндивидуальной вариабельности. Полиморфизм аллелей главного комплекса гистосовместимости (MHC), влияющий на аффинность и стабильность взаимодействия между молекулами MHC, антигенными пептидами и генами, кодирующими T-клеточный рецептор T-хелперов, может влиять на иммунный ответ и индукцию иммунной толерантности.
Иммунный ответ может развиваться даже в том случае, если последовательность аминокислот молекулы терапевтического белка полностью соответствует человеческой.
К другим генетическим факторам, влияющим на иммуногенность, может относиться полиморфизм генов, кодирующих цитокины, играющих роль в тонкой корректировке иммунного ответа (например, интерлейкин-10 (IL-10), трансформирующий фактор роста бета (TGF-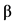 ) и др.).
) и др.).
Генетические факторы, обусловленные поломкой генов. Если терапевтический белок применяется для замещения эндогенного белка, низкие концентрации или даже отсутствие этого белка могут влиять на иммунологическую толерантность, поскольку у таких пациентов физиологический антиген может восприниматься как неоантиген.
Возраст. Данные, полученные в одной возрастной группе, не всегда можно экстраполировать на другие возрастные группы, поскольку иммунный ответ на введение терапевтического белка может зависеть от возраста. Иммунный ответ детей на белки может отличаться от иммунного ответа у взрослых. Если лекарственный препарат предназначен для лечения детей, необходимо провести исследования иммуногенности в этой возрастной группе (как это указано в подразделе 4.5.4 настоящей главы). Если препарат показан пожилым пациентам, следует учитывать вероятность изменения иммунного ответа у пациентов пожилого возраста.
Факторы, опосредованные заболеванием. Заболевание пациента само по себе может быть важным фактором развития нежелательного иммунного ответа.
У некоторых пациентов с хроническими инфекциями наблюдается большая склонность к развитию иммунного ответа, поскольку их иммунная система находится в активированном состоянии.
При других состояниях (например, дефиците питания, метастазировании опухоли, поздних стадиях ВИЧ-инфекции, органной недостаточности) развитие иммунного ответа на введение терапевтического белка менее вероятно из-за нарушенной функции иммунной системы.
В отношении некоторых препаратов известно, что возможность развития гуморального иммунного ответа может различаться в зависимости от показаний к применению или стадии заболевания. В связи с этим иммуногенность следует изучать в рамках клинических исследований отдельно для каждого заболевания или стадии его развития.
Сопутствующая терапия. Сопутствующая терапия может как снижать, так и повышать риск развития иммунного ответа на терапевтические белки. Обычно иммунная реакция на введение терапевтического белка снижается при применении иммунодепрессантов. Следует учитывать и предыдущее лечение, которое также может модулировать иммунный ответ на введение терапевтического белка и влиять на иммунную систему в течение длительного времени. Если клинические исследования проводились в комбинации с иммунодепрессантами, намерение применять терапевтический белок в монотерапии необходимо обосновать надлежащими клиническими данными по иммуногенности в отсутствии иммунодепрессантов, то есть данные об иммуногенности в комбинации с иммунодепрессантами не будут значимыми при принятии решения о возможности применения препарата в монотерапии.
Продолжительность, путь введения, методы лечения. К факторам, способным усилить иммунный ответ к терапевтическому белку, относятся путь введения, доза и режим дозирования.
Препараты, вводимые внутривенно, могут характеризоваться меньшей иммуногенностью, чем вводимые подкожно или внутримышечно.
При краткосрочном лечении вероятность развития иммунного ответа ниже, чем при долгосрочном; постоянное непрерывное применение реже вызывает развитие иммунного ответа, чем периодическое (нерегулярное) применение.
Нерегулярное лечение или повторное введение препарата после длительного перерыва может вызвать усиленный иммунный ответ.
Применение аналогичных или родственных белков в анамнезе. Ранее проведенная терапия аналогичными или родственными белками может вызвать предварительную сенсибилизацию организма и может быть причиной развития иммунного ответа. Некоторые белки при их использовании для заместительной терапии в прошлом могут вызывать выработку перекрестно реагирующих антител или развитие иммунологической памяти, которые будут влиять на последующее лечение.
4.1.2. Факторы риска иммуногенности, опосредованные лекарственным препаратом.
Факторы риска развития иммуногенности терапевтических белков биологического (биотехнологического) происхождения, опосредованные лекарственным препаратом, включают в себя происхождение и природу активной фармацевтической субстанции (структурная гомология белка, посттрансляционные модификации), модификацию нативного белка (например, пэгилирование), родственные и производственные примеси (например, продукты деградации, агрегаты, белки, липиды и ДНК клетки-хозяина), а также состав и лекарственную форму.
Структура белка. Аналоги эндогенных человеческих белков, полученные с использованием биотехнологических методов, могут быть причиной развития иммунного ответа в силу различий в последовательности аминокислот или изменений в структуре белка, возникших в результате посттрансляционных модификаций, физической, химической или ферментативной деградации и (или) модификации (например, дезамидирования, окисления и сульфатирования) возникающие на любом из этапов производственного процесса или при хранении. Особого внимания требуют гибридные белки, полученные из чужеродных и собственных белков, поскольку чужеродные компоненты могут провоцировать формирование иммунного ответа на собственный белок (распространение эпитопа, epitope-spreading). В таких случаях рекомендуется проводить идентификацию антигенного компонента гибридного белка. Гликозилирование - частая посттрансляционная модификация терапевтических белков, полученных с использованием биотехнологических методов. Такие модификации могут различаться по количеству и положению участков гликозилирования, последовательности, длине цепи и конформации олигосахаридных групп. В связи с этим, если один и тот же белок производится в различных условиях (например, изменение процесса культивирования клеток), характер посттрансляционных модификаций, а, следовательно, и иммуногенный потенциал белка могут изменяться. Это означает, что антитела, вырабатываемые в ответ на один лекарственный препарат, могут иначе реагировать на аналог, произведенный в измененных условиях, что необходимо учитывать при оценке иммуногенности.
Состав (рецептура). Состав (рецептуру) препарата подбирают в целях наилучшего поддержания нативной конформации терапевтического белка. Выбор оптимального и стабильного состава зависит от понимания физической и химической природы фармацевтической субстанции и вспомогательных веществ самих по себе и в комбинации друг с другом. Состав и происхождение вспомогательных веществ могут влиять на иммуногенность терапевтических белков, эти факторы необходимо рассматривать в качестве возможных причин подобных явлений. Их необходимо учитывать при изменении рецептуры.
На иммуногенный потенциал терапевтического белка могут также повлиять материал первичной упаковки и условия клинического применения, например, разведение в инфузионных растворах и использование инфузионного оборудования, произведенного из различных материалов.
Агрегация и образование аддуктов. Агрегация и образование аддуктов белков могут приводить к оголению новых эпитопов или к образованию поливалентных эпитопов, что в конечном счете может стимулировать иммунную систему. Факторы, от которых зависит агрегирование или образование аддуктов, включают в себя состав, процесс очистки, процедуры инактивации вирусов и условия хранения полуфабрикатов и готовых препаратов. Использование белков (например, альбумина) в качестве вспомогательного вещества может приводить к образованию более иммуногенных агрегатов. Важно постоянно контролировать содержание агрегатов и аддуктов в лекарственном препарате на протяжении всего его срока хранения (годности).
Примеси. Выделяют ряд примесей терапевтических белков, которые потенциально могут быть адъювантами. Белки клетки-хозяина из клеточного субстрата, прошедшие очистку наряду с фармацевтической субстанцией, могут вызывать иммунный ответ к себе. Однако возможно, что такие белки клетки-хозяина, липиды из клетки-хозяина или ДНК выступят в качестве адъюванта терапевтического белка.
4.2. Доклиническая оценка иммуногенности и ее последствий
Терапевтические белки в большинстве случаев характеризуются видовыми различиями, то есть белки человека воспринимаются организмом животного как чужеродные. В связи с этим прогностическая значимость доклинических исследований по оценке иммуногенности на животных считается низкой. Проведение доклинических исследований, направленных на прогнозирование иммуногенности у человека, обычно не требуется. При этом следует учитывать возможное появление в будущем новых технологий (новые модели in vivo, in vitro и in silico), которые могут оказаться полезными.
Тем не менее определение антител в доклинических исследованиях является обязательным элементом изучения токсичности при многократном введении с целью надлежащей интерпретации результатов таких исследований (в соответствии с требованиями глав 5.3 - настоящих Правил).
К тому же сравнение профиля антител с таковым препарата сравнения на животных моделях может являться частью исследований сопоставимости как для аналогичных (подобных) биологических лекарственных препаратов (как это указано в главах 15 - 15.11 настоящих Правил), так и при изменении процессов производства (как это указано в главах 9.1 - 9.2 настоящих Правил).
Иммунный ответ на терапевтический белок, являющийся аналогом эндогенного белка, может привести к появлению перекрестно реагирующих антител, направленных против эндогенного белка, если синтез эндогенного белка сохраняется. На животных моделях необходимо изучить имеющие значимость данные о последствиях индукции иммунного ответа к эндогенному белку или его отсутствие (дисфункцию). Необходимо учитывать реакции как гуморального, так и клеточного (если применимо) иммунитета. При отсутствии таких данных, но при наличии теоретических предпосылок о возможном риске по безопасности применения, для получения информации о возможных последствиях нежелательного иммунного ответа необходимо проведение исследований с иммунизацией животных терапевтическим белком или гомологичным белком соответствующего вида животных.
4.3. Разработка методов обнаружения и определения
иммунного ответа у человека
Нежелательная иммуногенность биологических лекарственных препаратов может проявляться в виде гуморального и клеточного иммунного ответа. Поэтому важно выбрать и (или) разработать методы и методологию оценки такого иммунного ответа. Основное внимание исследователей, как правило, направлено на выявление антител и их характеристику, поскольку это технически выполнимо и имеет большое значение для определения клинической значимости в плане безопасности и эффективности применения лекарственного препарата. Вместе с тем, клеточный иммунитет может также играть большую роль, заявитель должен рассматривать необходимость его оценки в индивидуальном порядке.
4.3.1. Стратегия количественного определения.
Выбор подходящей стратегии для оценки нежелательной иммуногенности биологических лекарственных препаратов имеет ключевое значение. Обычно стратегия должна включать в себя методы скрининга для идентификации образцов (пациентов) с наличием исследуемых антител, аналитические иммунохимические методы для подтверждения наличия антител и определения их специфичности и ряд функциональных методов для оценки нейтрализующей способности выявленных антител. Кроме того, необходимо предусмотреть другие аналитические методы, не направленные на выявление антител, например, методы определения имеющих значимость биомаркеров или фармакокинетических параметров (если применимо), которые позволят оценить и охарактеризовать влияние антител (других показателей иммунного ответа, если таковые выявлены (индуцированы)) на клинические эффекты. В соответствующих случаях у всех пациентов необходимо собирать исходные данные.
В приложении N 2 к настоящей главе приведен пример возможной стратегии выявления и установления характеристик антител.
4.3.2. Количественное определение антител.
Скрининговые методы количественного определения. Скрининговые методы количественного определения должны позволять обнаруживать антитела, выработанные в ответ на введение биологического лекарственного препарата, у всех серопозитивных пациентов (то есть во всех серопозитивных образцах). Следует учитывать, что неизбежно появление ложноположительных результатов, поскольку обычно абсолютной специфичности любого скринингового метода достичь невозможно, но вместе с тем ложноотрицательных результатов быть не должно. Желательными характеристиками скрининговых методов количественного определения являются чувствительность, специфичность, прецизионность, воспроизводимость и устойчивость.
Методы, подтверждающие наличие антител. Такие методы необходимы для исключения ложноположительных образцов (пациентов), отобранных в результате скрининга. Для этой цели могут быть использованы разные подходы, но при выборе метода следует учитывать ограничения и характеристики методов скрининга. Простое повторение скринингового метода количественного определения в его оригинальной форме для подтверждения специфичности, как правило, недостаточно и нецелесообразно.
Методы градации специфичности антител. В некоторых случаях необходимо использовать дополнительные методы, позволяющие получить подтверждающую информацию о специфичности выявленных антител. Эти данные способствуют подтверждению специфичности иммунного ответа.
Методы определения нейтрализующей активности. Для оценки нейтрализующей способности антител обычно используют биологические методы. При этом необходимо выбрать или разработать такой метод, который бы наилучшим образом соответствовал биологическому лекарственному препарату. Количественное определение с использованием биологического метода, применяемого для определения специфической активности (например, при определении качества серии при ее выпуске) как правило, можно использовать и для анализа нейтрализующих антител. Однако для оптимального определения нейтрализующей способности используемая методика зачастую требует доработки. При невозможности (недоступности) нейтрализующих методов количественного определения на основе клеток, допускается использовать методы конкурентного связывания с лигандом или другие альтернативы. Однако при их использовании необходимо подтвердить, что данные методы отражают нейтрализующую способность (активность) исследуемых антител.
4.3.3. Валидация методик.
Валидация аналитической методики является непрекращающимся процессом на протяжении всей разработки препарата. В целях проверки возможности достижения поставленных целей используемые в основных (опорных) исследованиях методики требуют валидации. С помощью валидационных исследований необходимо подтвердить, что методики достаточно линейны и дают концентрационно-зависимые отклики на изучаемые аналиты, а также имеют достаточную правильность, прецизионность, чувствительность, специфичность и устойчивость. Во избежание межлабораторной вариабельности в рамках основных (опорных) клинических исследований предпочтительным является использование централизованной лаборатории. На пострегистрационном этапе также необходимо учитывать потенциальную межлабораторную вариабельность.
Валидацию также необходимо проводить для подтверждения того, что эффект матрицы, обусловленный реактивами или веществами, содержащимися в образцах, не влияет негативным образом на полученные результаты. Это можно осуществить путем проведения исследований восстановления (recovery) и наблюдения за влиянием таких веществ, содержащихся в матрице, на ответ, полученный в отсутствие их влияния. Подобное исследование необходимо провести в отношении всего диапазона разведений проб, используемых в анализах, и, по меньшей мере, в некоторых случаях в отношении предельных разведений, которые можно достоверно оценить.
Остаточное содержание биологического препарата в крови пациента может образовывать комплексы с антителами к нему и вследствие этого снижать поддающуюся обнаружению концентрацию антител. Данное обстоятельство может различным образом влиять на методику в зависимости от ее разновидности, формата и типа антител. Существует несколько подходов к решению указанной проблемы, например, диссоциация иммунных комплексов кислотой, удаление избыточного количества биологического препарата путем твердофазной абсорбции, использование длительного периода инкубации и (или) использование методики, позволяющей проводить анализ в высоких разведениях. Сами подходы также подлежат валидации на применимость. Решение об их использовании принимается в индивидуальном порядке. В некоторых случаях указанную проблему можно преодолеть, осуществляя отбор проб для оценки профиля антител спустя достаточное количество времени после введения препарата, что позволяет ему элиминироваться из кровотока до отбора проб. Однако такой подход не должен значительно усложнять процесс обнаружения антител и лечение пациента.
Стандартизация и контроли. Аналитические методики необходимо стандартизовать, это требует подбора и (или) разработки соответствующих стандартных материалов, то есть использования соответствующих биологических стандартов и (или) хорошо описанных положительных и отрицательных контролей. Эти реактивы являются ключевыми для методики и необходимы для ее калибровки (градуировки) и валидации. Стандартизация особенно важна для методик, используемых при изучении нежелательной иммуногенности, поскольку она тесно связана с интерпретацией результатов анализа и дифференциацией между серопозитивными и серонегативными образцами.
4.3.4. Установление характеристик антител к терапевтическому белку.
При обнаружении у пациентов, получающих лечение, индуцированных антител необходимо установить их клиническую значимость. К этим исследованиям относятся иммунологическая и (или) биологическая оценка характеристик антител, а также изучение их влияния (других проявлений индуцированного иммунного ответа) на действие лекарственного препарата. Некоторые исследования могут быть проведены без использования анализа антител в рамках исследований in vitro, однако при таком подходе может потребоваться клиническое обследование пациентов, получающих лечение.
Установление характеристик антител. Если у пациента индуцирована выработка антител, образцы сыворотки или плазмы необходимо охарактеризовать с точки зрения содержания антител (концентрации (титра)), их нейтрализующей способности и других критериев, определяемых в каждом отдельном случае в зависимости от свойств биологического лекарственного препарата, целей исследования, особенностей пациентов, клинической симптоматики и других факторов. К ним могут относиться необходимость определения класса и подкласса (изотипа) иммуноглобулинов, аффинность и специфичность антител. В зависимости от цели исследования и этапа разработки препарата объем установления характеристик может быть различным. Используемые методики должны соответствовать своему целевому назначению.
Антитела, присутствие которых в положительных образцах подтверждено, должны быть исследованы в отношении их специфичности к действующему веществу (белковому компоненту), а также, если возможно, дифференцированы от антител, связывающихся с родственными и производственными компонентами. Известно, что антитела могут вырабатываться против всех и (или) любого из указанных веществ. Также желательно изучить перекрестную реактивность антител с другими лекарственными препаратами, содержащими сходные белки, и с их эндогенными аналогами (если это возможно и применимо).
Необходимо оценить нейтрализующую способность антител, обнаруженных в сероположительных образцах, поскольку обычно этот показатель коррелирует со снижением клинической эффективности биологического лекарственного препарата. В некоторых случаях имеет значение скрининг нейтрализующих образцов на наличие перекрестной нейтрализующей способности других лекарственных препаратов, содержащих такой же белок, или нейтрализации эндогенного белка, поскольку это может влиять на клиническую эффективность и безопасность применения исследуемого лекарственного препарата. Следует отметить, что нейтрализующая активность не обязательно соотносится с содержанием связывающих антител, т.е. образцы с высоким содержанием связывающих антител могут оказывать слабое нейтрализующее действие на биологическую активность, тогда как образцы с низким содержанием связывающих антител могут обладать различной степенью нейтрализующей способности (в зависимости от образца), которая зависит от вида лекарственного препарата и требует экспериментального изучения.
Стратегия оценки иммуногенности: дизайн и интерпретация. До начала оценки результатов клинических исследований необходимо тщательно и заранее продумать дизайн исследований иммуногенности, чтобы удостовериться, что в них включены все обязательные процедуры, а именно: выбор, оценка и установление характеристик аналитических методик, определение надлежащего режима отбора образцов, их объем, пробоподготовка и хранение, выбор статистических методов анализа данных. Это требуется в отношении методик, используемых для определения содержания антител и установления их характеристик, а также методов оценки клинического ответа в ответ на выработку антител (если они образуются). Указанные показатели необходимо определять в индивидуальном порядке, с учетом особенностей лекарственного препарата, пациентов и ожидаемых клинических параметров. Эти исследования могут быть источником ценной информации о наличии высокой иммуногенности к биологическим препаратам, ее свойствах и потенциальных клинических последствиях. Они необходимы при изучении сравнительной иммуногенности биоаналогичных (биоподобных) препаратов и изменении процесса производства зарегистрированных препаратов. Однако нежелательная иммуногенность может не обнаруживаться в рамках исследований, проводимых на предрегистрационном этапе, в силу ограниченного количества пациентов, в них участвующих. При этом необходимо продолжать оценку иммуногенности и ее клинический последствий на пострегистрационном этапе в рамках фармаконадзора. В некоторых случаях в целях оценки риска, обусловленного нежелательным иммунным ответом, необходимы пострегистрационные клинические исследования.
Дополнительные подробности о методах оценки и описания иммуногенности приведены в приложении N 1 к настоящей главе.
4.4. Возможные клинические последствия иммуногенности
4.4.1. Влияние на эффективность.
Факторы, которые определяют способность антител к терапевтическим белкам вызывать клинические последствия, включают в себя распознаваемый эпитоп, аффинность, класс антител и количество выработанных антител, а также фармакологические свойства биотехнологического лекарственного препарата. Кроме того, влияние на клинический результат может оказывать способность иммунных комплексов активировать комплемент и скорость их элиминации. Считается, что антитела, распознающие эпитопы терапевтического белка, не связанные с проявлением активности этого белка, как правило, вызывают менее выраженные клинические последствия. Тем не менее такие антитела могут влиять на фармакокинетику и тем самым косвенно влиять на эффективность. Нейтрализующие антитела, подавляющие биологическое действие препарата путем связывания с активным центром (или расположенными вблизи него центрами) или путем индукции конформационных изменений, могут быть причиной снижения эффективности. Должно быть проведено определение нейтрализующей способности антител в подтвержденных позитивных образцах с использованием соответствующих надежных аналитических методик (как это указано в подразделе 4.3 настоящей главы). Методы, используемые для определения нейтрализующей способности, должны позволять выявлять клинически значимые нейтрализующие антитела. Для определения зависимости между характеристиками антител и клиническим ответом требуется сравнение данных, полученных при оценке гуморального иммунного ответа, с результатами изучения образцов, взятых от пациентов, и результатами изучения клинического ответа (исхода). В большинстве случаев клинический эффект является специфичным для каждого отдельного препарата, например, увеличение популяции лейкоцитов под действием колониестимулирующих факторов либо повышение количества ретикулоцитов под действием эритропоэтина. Выбор такого типа исследований определяется свойствами лекарственного препарата и необходимостью их проведения. Во многих случаях бывает затруднительно идентифицировать клиническую конечную точку, достаточно чувствительную для подтверждения прямого влияния лекарственного препарата на клинический результат. Вариантом выхода из подобной ситуации может стать использование суррогатных конечных точек, например, фармакодинамических параметров или биомаркеров. Выбор подобных маркеров должен быть обоснован. Сравнение результатов лечения препаратом у пациентов до и после выработки антител может служить основанием для заключения о зависимости между выработкой антител (и их характеристиками) и клиническими результатами. Это может быть реализовано, либо путем внутригруппового анализа (ответ пациентов до и после формирования антител), либо путем сравнения с пациентами, участвующими в исследовании, у которых иммунный ответ не развился.
4.4.2. Влияние на безопасность.
Снижение эффективности и изменение профиля безопасности необязательно являются взаимосвязанными явлениями. Проблемы безопасности, например, реакции, обусловленные введением (инфузионные реакции), могут проявляться и в отсутствии изменения эффективности.
Немедленные последствия. У пациентов, в организме которых вырабатываются антитела, обычно развиваются реакции, обусловленные введением. Острые инфузионные реакции, включая анафилактические, могут развиваться во время введения (в течение нескольких секунд) или через несколько часов после инфузии. Заявители должны различать понятия "инфузионные реакции" и "анафилаксия", а также подробно описывать симптомы, подпадающие под определение "реакции, обусловленные введением". "Инфузионные реакции" - это симптомы, возникающие в тесной временной связи с введением препарата, они необязательно являются проявлением анафилаксии или реакции гиперчувствительности. Тем не менее острые реакции могут быть действительно аллергическими, а именно IgE-опосредованными реакциями типа I (анафилактические реакции), которые сопровождаются симптоматикой, включающей артериальную гипотензию, бронхоспазм, отек гортани или глотки, чихание и (или) крапивницу. Понятие "анафилаксия" должно применяться лишь в отношении таких проявлений, которые являются абсолютным противопоказанием к дальнейшему применению препарата. Однако большинство инфузионных реакций сопровождаются менее специфичной симптоматикой, для некоторых препаратов они чаще возникают при первом введении, иногда менее частые или тяжелые реакции наблюдаются при повторном введении. В таких случаях инфузионная реакция может не являться противопоказанием к дальнейшему применению препарата. К симптомам, обусловленным введением препарата, относятся головная боль, тошнота, повышение температуры тела, озноб, головокружение, "приливы", зуд, боли в грудной клетке или спине. Общеизвестно, что дифференциальная диагностика между инфузионными реакциями и анафилаксией может быть затруднительна, тем не менее это разграничение очень важно ввиду различных клинических последствий этих состояний.
Заявители должны уделять внимание не только инфузионным реакциям и симптомам анафилаксии, которые могут развиться в ответ на введение препарата, но и другим проявлениям, поскольку последствия иммуногенности зависят от индивидуальных свойств лекарственного препарата и могут сопровождаться неожиданными клиническими проявлениями.
Отсроченные последствия
Реакции гиперчувствительности замедленного типа иммунные комплексы. В дополнение к острым реакциям следует учитывать возможность развития гиперчувствительности замедленного типа (опосредованной T-клетками) и реакций, опосредованных иммунными комплексами. Риск развития таких реакций возрастает с увеличением интервала между введением лекарственного препарата. Такие реакции гиперчувствительности замедленного типа следует четко отличать от инфузионных реакций. Заявители должны обеспечить систематический сбор данных об отсроченных клинических последствиях применения терапевтического белка. К клиническим проявлениям таких реакций относят миалгию, артралгию с повышением температуры тела, кожную сыпь, зуд и т.д., однако необходимо осуществлять систематический сбор данных и о других, более редких клинических симптомах.
Помимо влияния на фармакологические свойства иммунные комплексы могут откладываться в тканях. Необходимо учитывать наличие сопутствующих заболеваний и критически оценивать потенциальное влияние иммунных комплексов на их течение, например, потенциальное ухудшение функции почек у пациентов с сопутствующей аутоиммунной патологией.
Перекрестная реактивность с эндогенными аналогами. Антитела, вырабатываемые в ответ на терапевтический белок, имеющий эндогенный аналог, могут перекрестно с ним реагировать, если образование эндогенного аналога сохранено (например, эритропоэтины). Частью предрегистрационной программы разработки препарата должно быть углубленное изучение гуморального иммунного ответа и перекрестного связывания антител в сочетании со строгим наблюдением за клиническими последствиями. Опыт применения других подобных лекарственных препаратов может быть полезен, но только его недостаточно.
Заявители, разрабатывающие лекарственные препараты нового поколения, такие как гибридные молекулы, совмещенные с физиологически функциональными молекулами, должны внимательно подходить к возможным последствиям перекрестной реактивности антител со всеми эндогенными компонентами.
4.5. Иммуногенность и клиническая разработка
4.5.1. Обоснование режима отбора образцов и кинетика гуморального иммунного ответа.
Оценка иммуногенности должна быть частью клинических исследований, поскольку высока значимость ее корреляции с клинической эффективностью и безопасностью применения препарата. При проведении клинического исследования необходимо оценивать иммуногенность у всех субъектов исследования, а не ограничиваться лицами с клинической симптоматикой (то есть обследовать только тех пациентов, у которых заподозрено изменение профиля безопасности или эффективности). В дополнение к запланированному периодическому отбору образцов у пациентов необходимо взять дополнительные образцы в случае возникновения каких-либо симптомов и при подозрении на развитие нежелательного иммунного ответа.
На развитие иммунного ответа на терапевтический белок влияют такие факторы, как доза, режим дозирования и тактика лечения (как это указано в подразделе 4.1 настоящей главы). В связи с этим режим отбора образцов для обнаружения иммунного ответа необходимо адаптировать и подобрать индивидуально для каждого препарата с учетом его характеристик, включая фармакокинетические свойства. Необходимо всегда исследовать образцы, полученные до введения лекарственного препарата. Поскольку необходимо оценивать общую частоту проявления иммуногенности для данного препарата по всем показаниям, желательно, чтобы режимы отбора образцов были сопоставимыми с указанными режимами в других клинических исследованиях, что позволит обеспечить возможность прямого сравнения частоты появления антител к препарату. Любые отклонения от этого принципа должны быть обоснованы. Заявители должны попытаться стандартизировать режимы отбора образцов, проведения анализа, определения и т.д., принимая во внимание опыт работы с подобными лекарственными препаратами. Во время лечения образцы всегда следует брать до очередного введения препарата, поскольку остаточное содержание действующего вещества в плазме может искажать результаты количественного определения (как это указано в подразделе 4.3 настоящей главы). Для обеспечения надлежащего качества исследуемого материала необходимо создать соответствующие условия хранения и транспортировки образцов.
Частота отбора образцов, а также сроки и объем проводимых анализов должны быть обоснованы, они зависят от степени риска, установленного в отношении конкретного лекарственного препарата, и возможных клинических последствий. В режиме необходимо предусмотреть регулярный отбор образцов и спланировать его таким образом, чтобы можно было отличить временно серопозитивных субъектов от пациентов со стойким образованием антител. Для определения общей иммуногенности препарата в заданных условиях необходимо учитывать результаты и тех и других. В частности, набольшее значение имеет стойкий иммунный ответ, поскольку у пациентов со стойким формированием антител более вероятно развитие клинических последствий, влияющих на эффективность и безопасность, тогда как транзиторный гуморальный иммунный ответ может в последующем разрешиться без дальнейших последствий.
В отношении препаратов, предназначенных для длительного применения при хронических заболеваниях, может потребоваться изучение динамики и продолжительности наблюдаемого иммунного ответа. Усилия исследователей должны быть направлены на сбор данных о возможных изменениях характеристик антител с течением времени, например, изменение не-нейтрализующей активности антител на нейтрализующую и по возможности у каждого конкретного пациента. При планировании клинической программы по изучению иммуногенности необходимо в каждом отдельном случае, например, при оценке степени риска, учитывать возможные последствия нежелательного иммунного ответа при долгосрочном применении. Более частый отбор образцов обычно практикуется на ранних этапах лечения, когда у пациентов обычно наблюдается самый высокий риск выработки антител. Поскольку долгосрочное лечение с большей вероятностью приводит к развитию иммунного ответа, в клинических исследованиях необходимо предусмотреть отбор образцов и на более поздних этапах лечения. Для препаратов, предназначенных для длительного непрерывного лечения, на предрегистрационном этапе необходимо получить данные по оценке иммуногенности в течение 1 года. Любые отклонения необходимо полностью обосновать, например, более короткой экспозицией или различным объемом данных в связи с несколькими путями введения. Если предусмотрены разные пути введения, на этапе подачи заявления о регистрации лекарственного препарата заявитель должен обосновать подход к оценке иммуногенности при всех путях введения. В зависимости от свойств лекарственного препарата и возможных рисков, обусловленных развитием нежелательного иммунного ответа, может потребоваться изучение достаточного количества экспозиций лекарственного препарата.
Для определения стойкости иммунного ответа по возможности необходимо провести анализ образцов, полученных после завершения терапии. Со временем содержание антител к терапевтическому белку у пациентов, у которых они изначально образовались, может снижаться, однако возможна обратная картина, например, если лекарственный препарат обладает иммунодепрессивными свойствами и за счет этого подавляет иммунный ответ, в том числе в отношении себя.
Если применимо, заявитель должен в составе регистрационном досье представить врачам инструкцию, как работать с пациентами, у которых снизилась эффективность, например, увеличить дозу, сократить интервал между введениями или прекратить лечение.
Результаты иммунологических исследований необходимо включить в общую характеристику лекарственного препарата.
4.5.2. Влияние на фармакокинетику лекарственного препарата.
Антитела, распознающие эпитопы, не связанные с активностью терапевтического белка, как правило, вызывают менее выраженные клинические последствия. Тем не менее такие антитела могут влиять на фармакокинетику и опосредованно на эффективность. "Элиминирующие" антитела могут быть нейтрализующими или не обладать такой активностью (не-нейтрализующие антитела), они могут снижать эффективность за счет удаления терапевтического белка из кровотока. В некоторых случаях не-нейтрализующие (связывающие) антитела могут даже повышать эффективность лекарственного препарата благодаря увеличению периода полувыведения действующего вещества или стимуляции механизмов его действия. Нейтрализующие антитела могут инактивировать лекарственный препарат, как за счет повышения его клиренса, так и без вовлечения этого механизма. При необходимости снижение эффективности может быть описано в соответствии с принципами стратегии анализа, приведенными в подразделе 4.3 настоящей главы. Изменение фармакокинетики может быть ранним признаком формирования антител. При обнаружении антител во время проведения исследований в соответствии с клинической программой необходимо изучать их возможное влияние на фармакокинетику лекарственного препарата (см. также документ Союза по клиническим исследованиям фармакокинетики терапевтических белков).
4.5.3. Методологические аспекты оценки сопоставимости иммуногенного потенциала как элемента сравнительных исследований.
Внесение изменений в процесс производства может повлиять на иммуногенность лекарственного препарата. При изменении процесса производства зарегистрированного лекарственного препарата проводят поэтапные исследования сопоставимости (в соответствии с требованиями глав 9.1 и 9.2 настоящих Правил). Если результаты первичных физико-химических и биологических испытаний указывают на различия между лекарственными препаратами, полученными до и после внесения изменений в процесс производства, необходимо рассмотреть возможные последствия внесенных изменений на показатели безопасности и эффективности, включая влияние на иммуногенность. Даже если при первичных физико-химических и биологических испытаниях различия не выявлены, необходимо принимать во внимание изменение иммуногенности вследствие невыявленных причин. Объем исследований иммуногенности при их необходимости должен быть определен на основании анализа рисков с учетом характера наблюдаемых различий, возможного влиянии на клиническую картину заболевания, а также опыта, ранее накопленного при разработке данного лекарственного препарата и для лекарственных препаратов этого класса. Выбор соответствующей категории пациентов необходимо осуществить таким образом, чтобы наилучшим образом проследить все различия, не ограничиваясь только оценкой показателей иммуногенности. В целях проведения таких сравнений заявителям необходимо предпринять усилия для выбора однородной и клинически подходящей популяции пациентов. Ввиду ожидаемых различий в восприимчивости, данные об иммуногенности, полученные от здоровых добровольцев, не являются подходящей альтернативой. В целях подтверждения того, что изменение процесса производства не повлияло на эффективность и безопасность, иммуногенность большинства лекарственных препаратов изучают в рамках клинического исследования у пациентов ранее не получавших лечения. Оценку иммуногенности как части клинического исследования при изучении сопоставимости предпочтительно проводить путем прямого сравнения лекарственного препарата, полученного до изменения процесса производства и после него. Необходимо использовать те же аналитические методики.
Изменения иммуногенности в результате изменения процесса производства могут потребовать специальной стратегии и обновления плана по управлению рисками (в соответствии с требованиями подраздела 4.6 настоящей главы). При наличии риска развития редких иммуноопосредованных нежелательных явлений их можно оценить после внесения изменений на этапе пострегистрационного применения препарата.
4.5.4. Иммуногенность у детей.
Терапевтические белки применяются у детей. Известно, что иммунный ответ ребенка отличается от иммунного ответа взрослого человека.
При изучении препарата у детей тщательному выбору и обоснованию подлежат режим дозирования и тактика лечения. По возможности результаты исследований следует анализировать по возрастным группам, а данные по иммуногенности - оценивать и представлять отдельно для каждой возрастной группы, что позволит выявить потенциально уязвимые группы.
В отношении заместительной терапии рекомбинантные технологии позволили разработать белки, применяемые при генетических нарушениях. Наиболее вероятными субъектами клинических исследований таких лекарственных препаратов являются дети, которые могут быть подвержены повышенному риску образования антител.
4.6. План управления рисками
В состав регистрационного досье должен быть включен план управления рисками в соответствии с правом Союза в сфере обращения лекарственных средств, рекомендациями по фармаконадзору, включая правила надлежащей практики фармаконадзора Союза, утверждаемые Комиссией. Вопросы иммуногенности всегда должны быть предметом рассмотрения в плане по управлению рисками (далее - ПУР) с учетом всех рисков, выявленных во время разработки лекарственного препарата, а также потенциальных рисков и последствий нежелательного иммунного ответа у пациентов. Описание рисков и деятельность, направленная на минимизацию рисков, должны соответствовать принципам, изложенным в данной главе. Следует подчеркнуть, что оценка иммуногенности основывается на междисциплинарном подходе и требует обязательного участия специалистов по качеству, доклиническим и клиническим исследованиям.
Объем данных по иммуногенности, которые могут быть получены в ходе программы клинической разработки биотехнологического лекарственного препарата до его регистрации, зависит от частоты развития явлений, обусловленных иммуногенным потенциалом белка и частотой встречаемости заболевания. Таким образом, после регистрации может понадобиться дальнейшее систематическое изучение иммуногенности, что необходимо отразить в плане управления рисками.
Объем данных об иммуногенности, который необходимо собрать на пострегистрационном этапе, зависит от различных факторов, включая:
факторы, опосредованные заболеванием, такие как распространенность, восприимчивость пациентов, доступность альтернативных способов лечения, продолжительность лечения и др.;
данные по иммуногенности, полученные до регистрации, включая влияние на эффективность и безопасность;
данные об опыте применения аналогичных белков или членов того же класса с оценкой проявлений иммуногенности, включая белки, полученные с помощью аналогичных производственных процессов;
серьезность иммунных реакций.
Однако белки, полученные с использованием биотехнологических методов, следует рассматривать индивидуально, и следовательно возможность экстраполяции данных, полученных на других сходных белках, можно использовать ограниченно и только с полным обоснованием.
В последующем иммуногенность можно изучать на пострегистрационном этапе, например, путем усиленного сбора данных о возможных иммуноопосредованных нежелательных явлениях (включая снижение эффективности), или при проведении фармакоэпидемиологических исследований.
Поскольку систематический отбор образцов на пострегистрационном этапе затруднителен, необходимо уметь выявлять потенциальные нежелательные иммунные реакции на основании подозрительных сигналов о безопасности и (отсутствии) эффективности. Данное обстоятельство требует заблаговременного отражения в ПУР оценки таких явлений. Держатель регистрационного удостоверения должен разработать стандартизированный алгоритм подробного расследования случаев, подозрительных в отношении иммунного ответа, включая методы подтверждения образования антител.
План по управлению рисками должен включать следующее:
идентификацию и описание рисков (например, описание случаев, методики количественного определения антител);
мониторинг рисков (например, особые модели, выявляющие взаимосвязь между риском и лекарственным препаратом);
стратегии минимизации и ослабления рисков (например, при необходимости - возможность использования только внутривенного введения, мероприятия при обнаружении риска и др.);
оповещение по вопросам рисков (например, сообщение врачам и пациентам о способах минимизации и ослабления рисков, распространение среди врачей информации об аналитических системах и специфичных методах анализа для выявления антител);
мониторинг мероприятий, необходимых для обеспечения эффективной минимизации рисков.
Заявители должны учитывать новые данные об иммуногенности путем принятия соответствующих мер, например, внесения сведений по иммуногенности в информацию о лекарственном препарате, обновления ПУР и других мероприятиями по минимизации риска (например, образовательными программами и т.д.). При планировании оценки иммуногенности на пострегистрационном этапе в большинстве случаев применимы указания настоящей главы.
При изменении процесса производства последствия влияния такого изменения на иммуногенный потенциал лекарственного препарата необходимо отразить в ПУР.
Приложение N 1
к главе 11 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ К ХАРАКТЕРИСТИКЕ МЕТОДОВ И ОЦЕНКЕ ИММУНОГЕННОСТИ
I. Разновидности количественного определения антител
1. Методы скрининга
Необходимость скрининга относительно большого количества образцов требует использования высокопроизводительных аналитических методик с высокой степенью автоматизации. К этим методам относятся иммунологические методы, а также радиоиммунопреципитация и поверхностный плазмонный резонанс. Все они направлены на выявление взаимодействия между антигенами и антителами (связывание), но основаны на разных научных (технических) принципах.
Иммунологические виды анализа представляют большую группу методик и основаны на разнообразных способах и системах обнаружения. К ним относятся аналитические методики, выявляющие прямое связывание, связующие методики, методики захвата (сэндвич-анализ) и конкурентные иммунологические анализы, основанные на радиолигандных, ферментных, флюоресцентных, хемилюминесцентных и электрохимических люминесцентных системах обнаружения.
2. Методы, подтверждающие наличие антител
Для этих целей можно использовать различные аналитические методики, ввиду меньшего по сравнению с методами скрининга, количества образцов, подлежащих анализу, здесь высокая производительность играет меньшую роль. В целях подтверждения специфичности, необходимо включать методики, позволяющие ее оценить. Например, добавление избыточного количества антигена в образец до начала работы с методиками связывания, приводит к абсорбции антител и, как следствие, снижению частоты положительных сигналов от истинно серопозитивных образцов. Выявление в качестве анализируемого вещества иммуноглобулинов в некоторых аналитических методиках, например, путем использования антииммуноглобулиновых реактивов, может помочь в обнаружении доподлинно серопозитивных образцов.
В некоторых сложных случаях рекомендуется использовать аналитические методики, основанные на иных научных (технических) принципах, чем используемые в методах скрининга. Кроме того, следует учитывать различие характеристик используемых методик, например, их различную чувствительность.
3. Методы градации специфичности антител
Для дифференцирования специфичности выявленных антител используют аналитические иммунологические методы, такие как иммуноблоттинг и радиоиммунопреципитация.
4. Методы определения нейтрализующей активности
В зависимости от свойств лекарственного препарата для определения нейтрализующей активности антител могут быть использованы биологические методы количественного определения, включая функциональные.
Обычно для анализа выбирают определенную концентрацию биологического лекарственного препарата, а его разведения используют для оценки ингибирующего действия при снижении концентрации антител. Это позволяет определить нейтрализующую дозу и рассчитать нейтрализующую способность (титр) антител для каждого образца.
5. Методы оценки клеточного иммунного ответа
Стратегия оценки клеточного иммунного ответа, обусловленного биологическим лекарственным препаратом, как правило, сложнее по сравнению с оценкой гуморального ответа. Если такие испытания необходимы, требуются их специальная разработка и выбор метода для каждого конкретного случая индивидуально. В большинстве случаев развитие эффективного ответа с участием IgG обусловлено участием антигенспецифичных T-хелперов.
Примерами методов количественного определения, используемых для обнаружения (оценки) клеточного иммунного ответа, являются методы определения стимуляции (пролиферации) T-клеток, а также методы оценки выработки (высвобождения) цитокинов (например, IL-2, IL-4, ИФН-гамма). Это предполагает использование T-клеток, иногда совместного культивирования T-клеток с клетками других популяций, например, дендритными клетками. Для этих целей обычно используют метод иммуноферментных пятен (Elispot) и проточную цитометрию. В некоторых случаях используют анализ клеточно-опосредованной цитотоксичности.
Иногда необходимо использовать более детальные исследования, включающие оценку клеточного иммунного ответа. Изучение B-клеток памяти (некоторых случаях T-клеток памяти) может дать полезную информацию о природе иммунного ответа, результаты исследования также могут иметь прогностическую ценность в отношении потенциальной иммуногенности исследуемого лекарственного препарата. С этой целью могут быть проведены исследования с использованием пептидов или белков (в зависимости от вида анализа и его целей), и использован метод иммуноферментных пятен. В других случаях могут быть полезны более сложные исследования клеточного иммунитета, например, изучение регуляторных T-клеток. В зависимости от целей и задач, необходимость в таких исследованиях должна рассматриваться индивидуально.
II. Характеристики аналитических методов
Выбор, оптимизацию и анализ результатов испытаний следует выполнять в соответствии с поставленными задачами. Важность и требования к характеристикам методов (перечень некоторых из них приведен выше в подразделе "Методы скрининга") зависят от используемой методики. Например, может отсутствовать необходимость в высокой чувствительности метода, если его не используют для определения содержания антител, выработанных в ответ на лечение определенным биологическим лекарственным препаратом. Нецелесообразно разрабатывать и использовать высокочувствительные методы для определения таких антител, особенно если чувствительность метода достигается в ущерб другим важным характеристикам, например, специфичности, устойчивости.
Выбор самого простого метода, удовлетворяющего всем требованиям, является разумным подходом, особенно при высокой значимости большой производительности, например при скрининге. Тем не менее следует внимательно следить за тем, чтобы это отрицательно не сказывалось на других этапах оценки иммуногенности. Например, прямой иммуноферментный анализ (ИФА) с антигеном, иммобилизованным на поверхности лунок планшета, представляет простейший вид испытания, но характеризуется большим количеством ложноположительных результатов. Кроме того, для этих методов характерна высокая частота ложноотрицательных результатов при использовании образцов, содержащих низкоаффинные антитела или определенные их изотипы или подклассы. Во избежание вышеуказанных проблем необходимо выбрать более подходящий вид анализа, например "связующие" методы, электрохемилюминесценцию или поверхностный плазмонный резонанс. Могут встречаться ложноотрицательные результаты скринингового метода, обусловленные маскировкой эпитопов. Во избежание этого следует придерживаться соответствующей стратегии, например, использовать методы, при выполнении которых специфическая маскировка определенных эпитопов невозможна.
III. Стандартизация, стандартные материалы, хорошо
охарактеризованные контроли и валидация методик
Для всех видов методик необходимы серопозитивные стандартные образцы (материалы, контроли). Они используются для подтверждения отклика методики и ее калибровки (градуировки). По возможности, они должны являться препаратами крови человека с высоким содержанием антител, полученными в достаточном количестве для продолжительного использования. Их свойства должны быть хорошо охарактеризованы; хранение таких материалов осуществляется в надлежащих условиях (в виде лиофилизата или замороженными при подходящих температурах). Стандартные препараты для биологических методов количественного определения, направленных на определение нейтрализующей активности, должны обладать высокой нейтрализующей способностью, однако для проведения валидации, необходимо располагать препаратами антител, не обладающими нейтрализующей активностью.
В ряде случаев для приготовления необходимых стандартных препаратов в наличии может оказаться не достаточное количество плазмы человека. В этой ситуации наилучшим подходом, который позволяет избежать проблем в силу особенностей определенного донора, является объединение образцов. Иногда объема человеческой плазмы не только не хватает для объединения, она может вообще отсутствовать, например, на ранней стадии разработки, в таких случаях единственно возможным решением является использование плазмы животных. При этом требуется тщательный выбор вида животных, а применение такой плазмы носит ограниченный характер по сравнению со стандартными препаратами, полученными из крови человека. Например, иммунохимические анализы, включающие использование антител к человеческим иммуноглобулинам, могут давать ложные результаты при инкубации с нечеловеческими антителами, а результаты любых исследований могут отличаться от результатов, полученных при изучении антител человека, содержащихся в человеческой плазме.
В силу гетерогенности структуры, специфичности и авидности иммуноглобулинов, содержащихся в стандартах образцах и пробах, калибровка (градуировка) иммунологических методов анализа является трудной задачей. Это обуславливает сложность и невозможность прямого правильного сравнения испытуемых образцов и стандартных материалов, особенно по их массе. Вследствие чего калибровку таких методов анализа необходимо осуществлять, используя хорошо описанные приемлемые обоснованные подходы. Одним из способов может служить представление результатов иммунологических методов анализа в виде титра, основанное на стандартных процедурах вычисления его значения. Другим способом является вычисление относительной концентрации антител в испытуемых образцах и положительных контролях.
Биологические методы анализа, используемые для оценки нейтрализующей способности антител, необходимо калибровать с помощью международных стандартных образцов (стандартных препаратов) (при наличии). Это позволяет выразить нейтрализующую активность в понятных единицах биологической активности лекарственного препарата (препарата крови) и получить необходимую информацию для валидации аналитических методик. При недоступности указанных стандартов необходимо разработать собственные препараты. Во многих случаях рекомендуется выражать нейтрализующую способность образцов по их объему, необходимому для нейтрализации постоянной биологической активности препарата, например, в миллилитрах плазмы или заранее заданных единицах биологической активности биологического препарата. В иных случаях допускается использовать разведение образцов (титр), необходимое для нейтрализации биологической активности препарата.
Также настоятельно рекомендуется располагать набором стандартных материалов, содержащих различные количества антител, и набором антител с различными свойствами, которые можно использовать для установления характеристик и валидации аналитических методик и которые могут служить показателями их функциональности. По возможности они должны включать 1 или более препаратов с низким содержанием антител (близким к нижнему пределу обнаружения) и препаратов антител с низкой авидностью.
В целях определения исходных параметров аналитической методики, установления характеристик и валидации необходимы негативные стандартные образцы и их контроли. Исходные параметры аналитической методики у здоровых субъектов можно с легкостью определить путем определения ее результатов при анализе образцов, полученных от достаточного числа таких субъектов и их анализа с целью получения статистически надежных исходных значений. Однако этот способ может не позволить охарактеризовать исходные параметры аналитической методики при анализе образцов, полученных от пациентов, поэтому такие параметры придется определять отдельно, используя образцы от пациентов, полученных до начала лечения, или образцы, полученные от других подходящих субъектов. Как бы то ни было, в образцах некоторых субъектов (пациентов) могут содержаться антитела, выработанные еще до начала лечения, или другие вещества, которые могут давать значительные ложноположительные результаты, поэтому в целях обеспечения правильной интерпретации результатов, полученных после начала лечения, необходим скрининг пациентов.
Необходимо установить требования и подобрать приемлемые спецификации для используемых в анализах реактивов, по меньшей мере важнейших.
IV. Интерпретация результатов
Необходимо установить четкие критерии разделения образцов на серопозитивные и серонегативные, а также критерии подтверждения положительного результата. Подходы к решению этой задачи могут быть разными в зависимости от метода и других факторов, что требует должного обоснования. Общим способом определения порога принятия результата как положительного при выполнении иммунологических методов является установление исходных параметров. Для определения значения такого порога рекомендуется использовать статистические методы. С другой стороны, допускается использование реальных данных (например, удвоенное значение, установленное при изучении исходных параметров) в качестве минимального положительного значения.
Приложение N 2
к главе 11 Правил
проведения исследований
биологических лекарственных средств
Евразийского экономического союза
ПРИМЕР СТРАТЕГИИ ВЫЯВЛЕНИЯ И ХАРАКТЕРИСТИКИ АНТИТЕЛ
┌──────────────────────────────────────────────────────────────────┐
│ Отбор образцов пациентов в определенных временных точках │
└──────────────────────────────────┬┬──────────────────────────────┘
││
\/
┌───────────────────────┐ ┌────────┐ ┌───────────┐
│"-" образцы отсеиваются│/───│Скрининг│───\│"+" образцы│
└───────────────────────┘\───└────────┘───/└───────────┘
/\ ││
││ ┌─────────────────────┐ ││
│└──────────│Подтверждающий анализ│/─────┘│
└───────────└─────────────────────┘\──────┘
││
\/
┌─────────────┐ ┌─────────────────────┐ ┌─────────────┐
│Биологический│/───│ Подтвержденные "+" │───\│ Установление│
│ анализ │\───│ образцы │───/│характеристик│
└─────────────┘ └─────────────────────┘ └─────────────┘
││ ││
\/ \/
┌──────────────────────────────────────────────────────┐
│Оценка корреляции между охарактеризованными антителами│/───┐
│ и клиническим ответом на биологический препарат │\──┐│
└──────────────────────────────────────────────────────┘ ││
││
┌──────────────────────────────────────────────────────┐ ││
│ Количественное определение клинических маркеров │───┘│
│ и оценка клинического ответа у пациентов │────┘
└──────────────────────────────────────────────────────┘
"-" - отрицательный
"+" - положительный
Глава 12. Оценка иммуногенности препаратов моноклональных
антител, предназначенных для применения в клинической
практике in vivo
В настоящей главе рассматриваются вопросы, связанные с проявлениями нежелательной иммуногенности препаратов моноклональных антител (далее - МкАТ), предназначенных для клинического применения. К ним относятся факторы, влияющие на иммуногенность МкАТ, клинические последствия иммуногенности, аналитические проблемы, оценка нейтрализующих антител на моноклональные антитела и вопросы подхода, основанного на рисках, к анализу иммуногенности МкАТ.
1. Введение
Проявление нежелательной иммуногенности может быть значительной проблемой при лечении пациентов биологическими лекарственными препаратами. Рекомендации по оценке иммуногенности лекарственных препаратов на основе белков, полученных с использованием биотехнологии, представлены в главе 11 настоящих Правил, которые применимы и к лекарственным препаратам МкАТ. Несмотря на то, что многие аспекты иммуногенности МкАТ не отличаются от аспектов иммуногенности других терапевтических белков, некоторые из них требуют более пристального рассмотрения. Индукции моноклональными антителами антител, которые перекрестно реагировали и нейтрализовали бы эндогенные антитела (как, например, в случае с эритропоэтином), не ожидается, поскольку их не применяют в качестве заместительной терапии. Чаще всего лекарственные препараты МкАТ применяются в качестве терапевтических или диагностических средств при наличии терапевтической или диагностической альтернативы. Тем не менее некоторые специфичные аспекты иммуногенности характерны исключительно или преимущественно для препаратов МкАТ или новых препаратов на основе модифицированных МкАТ (например, Fab-фрагментов, scFv-одноцепочечных Fv-фрагментов, нанотел, миниантител), которые рассматриваются в настоящей главе.
Препараты МкАТ представляют значительную и очень важную подгруппу биологических лекарственных препаратов. Диапазон показаний к применению МкАТ при лечении заболеваний очень широк. Применение многих препаратов МкАТ сопровождается проявлениями нежелательной иммуногенности, в некоторых случаях это приводит к неполноценному клиническому ответу или развитию редких серьезных нежелательных реакций, которые требуют клинического вмешательства. Широкий спектр разрабатываемых и зарегистрированных по различным показаниям к применению препаратов МкАТ препятствует составлению частных рекомендаций, применимых во всех ситуациях.
2. Область применения
Общие принципы касаются вопросов разработки и проведения систематизированной оценки нежелательного иммунного ответа у реципиентов после введения им терапевтического или in vitro диагностического МкАТ. Требования относятся к препаратам МкАТ, их производным (например, Fab фрагменты, ScFv, наноантитела, миниантитела) и продуктам, содержащим компоненты МкАТ (например, конъюгаты, Fc-связанные гибридные белки).
В настоящей главе рассматриваются основные аспекты качества и клинические проявления, имеющие важное значение для адекватного решения проблем выявления и оценки риска развития нежелательного иммунного ответа на применение конкретного препарата МкАТ у пациентов с определенным заявленным показанием к применению.
Положения, приведенные в настоящей главе, распространяются на препараты, находящиеся на завершающей стадии разработки, в частности, на этапе подачи заявления о регистрации, однако многие положения применимы и к более ранним этапам разработки препаратов МкАТ.
3. Общие положения
Настоящую главу следует рассматривать в совокупности с другими главами настоящих Правил и иными соответствующими актами, входящими в право Союза.
4. Проблемы скрининговых и подтверждающих исследований,
используемых при оценке иммуногенности препаратов
моноклональных антител
4.1. Аналитические методы обнаружения антител
Для определения содержания антител к препарату МкАТ могут быть использованы любые форматы иммунологических методов количественного определения. Вместе с тем методы количественного определения, используемые для обнаружения антител к МкАТ, чаще вызывают затруднения, являются более сложными и иногда технически трудновыполнимыми. Многие стандартные форматы количественного определения включают использование анти-иммуноглобулиновых реагентов, в том числе, протеина A или протеина G, антител к иммуноглобулинам, однако они неприменимы для обнаружения антител к МкАТ, поскольку часто связываются с самим препаратом. Таким образом, простые методы, например, ИФА или радиоиммунная преципитация непригодны для МкАТ, если только их не приспособили к преодолению указанного затруднения. В связи с этим необходимо разработать другие подходы к определению МкАТ. Общий подход состоит в использовании "связующего" формата, например, для ИФА или электрохемилюминесценции (далее - ЭХЛ), которые не требуют анти-иммуноглобулиновых реактивов, и поэтому могут напрямую использоваться в исследованиях с МкАТ. В некоторых случаях этот метод может быть менее чувствительным, чем другие иммунологические методы, и требовать существенных усилий при разработке, чтобы создать подходящий метод количественного определения. Он также недостаточно эффективно обнаруживает образующиеся в некоторых случаях IgG4-антитела. Другим подходом является использование метода поверхностного плазмонного резонанса (далее - ППР). Он не требует использования антииммуноглобулиновых реактивов для обнаружения антител к МкАТ. Этот метод идет в реальном времени, вследствие чего он быстр и позволяет обнаруживать быстро диссоциирующиеся антитела, которые могут быть упущены другими методами. Поскольку ППР просто обнаруживает связывание белка с покрытым чипом, необходимо подтвердить, что сигнал исходит от антител. Он может быть менее чувствительным по сравнению с другими методами обнаружения высоко аффинных антител и в отсутствие автоматизированной системы пробоподготовки может иметь низкую производительность (низкий выход).
Пробы (как правило, сыворотка или плазма) могут содержать вещества, способные искажать результаты анализа, то есть вызывать эффект матрицы, заключающийся в получении ложноположительных или ложноотрицательных результатов и (или) неправильной оценке содержания антител.
4.2. Наличие препарата моноклональных антител
в образцах для анализа
Интактные препараты МкАТ имеют относительно длительные периоды полувыведения и сохраняются в кровотоке в течение длительного времени. Даже их фрагменты могут находиться в крови в течение нескольких дней. Это может существенно осложнить обнаружение иммунного ответа вследствие наличия препарата МкАТ в пробах, собранных в целях обнаружения антител. Это, как правило, приводит к артефактно низкой оценке содержания антител в соответствующих (пораженных) пробах и может быть настолько выраженным, что может привести к ложноотрицательным результатам. Предложено несколько подходов преодоления указанных сложностей. Первый подход заключается в том, чтобы отложить отбор проб до снижения содержания препарата МкАТ до значений, не вызывающих затруднений. Этот подход позволяет решить проблему некоторых препаратов МкАТ, но требует тщательного изучения, поскольку может привести к невыявлению иммуногенности вследствие снижения содержания индуцированных антител до необнаруживаемых количеств к моменту отбора проб. Другой подход заключается в использовании методологии, которая наименее подвержена влиянию указанной проблемы. Представляется, что методики, основанные на ЭХЛ, гораздо меньше подвержены влиянию остаточного содержания препарата в пробах, чем другие методы, включая стандартные связующие ИФА. Широко описываемой методикой решения проблемы является включение в план исследования предварительного этапа диссоциации комплекса антиген-антитело, чтобы разрушить все комплексы перед определением антител. Описаны различные версии методик, включая кислотную инкубацию, в некоторых случаях вместе с аффинным разделением препарата, однако следует с осторожностью анализировать их результаты, поскольку дополнительные этапы могут приводить к невалидности методики. В рамках третьего подхода можно подвергнуть пробы разведению, чтобы добиться остаточного содержания препарата, не влияющего на методику. Этот подход требует соблюдения большой осторожности, поскольку может привести к ложноотрицательному заключению об иммуногенности вследствие недостаточной чувствительности обнаружения антител в разведенных пробах с помощью такой методики. В некоторых случаях остаточное содержание МкАТ в пробах необходимо подвергать количественному определению.
Во многих случаях в целях снижения искажающего влияния препарата при разработке метода обнаружения антимоноклональных антител, его валидации и испытании используется комбинация всех трех подходов.
4.3. Подтверждающие анализы
Подтверждающие методы количественного определения подвержены тем же проблемам, что и скрининговые. Необходимо подобрать правильный подтверждающий метод количественного определения, принимая во внимание использованный скрининговый метод. Допускается использование протеина A и протеина G в подтверждающих методах для того, чтобы подтвердить, что положительный результат действительно обусловлен иммуноглобулином, однако в этих целях допускается использовать и другие подходы.
4.4. Контрольные образцы
Ключевой проблемой исследований иммуногенности МкАТ является наработка сывороток, которые будут служить положительным контролем. Выбранная сыворотка, являющаяся положительным контролем, или очищенное антитело необходимы для мониторинга чувствительности и специфичности метода количественного определения. Если сыворотку человека получить невозможно (например, на ранних фазах разработки препарата), то единственным выходом остается использование сыворотки животных. Выбор видов животных для этих целей приводит к важным последствиям. Нечеловекообразные приматы вырабатывают выраженный анти-CDR и антикаркасный ответ на человеческие и гуманизированные МкАТ, что может очень сильно имитировать ответ организма человека и служить подходящим положительным контролем. Тогда как неприматы вырабатывают антитела преимущественно к константным участкам МкАТ, что не характерно для иммунного ответа человека. В некоторых случаях положительным контролем может служить использование антиидиотипической антисыворотки или МкАТ. Необходимо подобрать правильные отрицательные контроли. В целях подтверждения специфичности подтверждающих методов количественного определения возможно использование проб, содержащих нерелевантные МкАТ.
5. Оценка нейтрализующей способности антител,
индуцированных лекарственным препаратом
моноклональных антител
Свое действие МкАТ оказывают с помощью различных механизмов, начиная от простого связывания с антигеном, которое само по себе опосредует клинический эффект, до связывания с антигеном и опосредуя 1 или несколько иммунобиологических механизмов, которые совместно определяют совокупный клинический ответ. Следовательно, несмотря на то, что может показаться, что простое связывание является единственным механизмом, определяющим клиническую эффективность, свой вклад могут вносить также и другие эффекты. В некоторых случаях множественные функции МкАТ действуют аддитивно или синергично, приводя к совокупному комбинированному клиническому эффекту, что в некоторых случаях поддается трудной экспериментальной дифференциации, позволяющей установить, каким образом МкАТ оказывает свое клиническое действие. В связи с этим при использовании интактных МкАТ необходимо с осторожностью выдвигать предположение о том, что Fc-опосредованные иммунобиологические эффекты препарата не вносят вклад в клиническую эффективность, даже если простое связывание с антигеном рассматривается в качестве основного механизма действия. В этой связи преимуществом для определения нейтрализации обладает количественное определение на основе клеток. В таких случаях, используя биологические и иммунологические методы количественного определения, необходимо провести тщательное установление биологических характеристик МкАТ. Затем необходимо оценить свойства МкАТ, чтобы подобрать надлежащую стратегию количественного определения нейтрализации.
Антитела, нейтрализующие биологическую активность биологических препаратов, способны снижать их клиническую эффективность. Необходимо определять нейтрализующую способность всех выработанных антител. Отсутствие таких данных требует обоснования. В отношении большинства биологических препаратов наиболее подходящим методом количественного определения нейтрализующей способности антител является количественный биологический метод, определяющий нейтрализацию антителами биологической активности препарата. Вместе с тем характер клинического способа действия МкАТ предполагает, что наиболее выражено снижают клиническую эффективность выработанные антитела, блокирующие связывание МкАТ с мишенью. Таким образом, методами выбора с целью определения нейтрализующей способности МкАТ являются конкурентные методы связывания с лигандом, а не классические количественные биологические методы. Это отличает МкАТ с точки зрения оценки иммуногенности от других классов биологических препаратов.
6. Управление рисками иммуногенности препаратов
моноклональных антител
6.1. Идентификация рисков
Иммуногенность МкАТ является сложным явлением: имеется ряд трудно понимаемых факторов, которые затрудняют точное прогнозирование клинически значимого иммунного ответа на терапевтическое или диагностическое моноклональное антитело. Разработаны in vitro доклинические подходы, нацеленные на обнаружение образованных T-клеточных эпитопов, однако они обладают ограниченной способностью прогнозировать иммуногенность препарата у человека. Вместе с тем такие методики могут пригодиться при выборе молекул-кандидатов для дальнейшей разработки.
Как указано в главе 11 настоящих Правил, необходимо изучить стандартные аспекты иммуногенности каждого нового МкАТ для медицинского применения, учитывая его свойства, характер предлагаемого применения и показание к применению.
В основе планирования будущих исследований лежат предварительные данные об иммуногенности, полученные по результатам ранних клинических исследований, например, изучение функциональных характеристик биоаналитических методик, обнаружение предсуществующих антител или иные факторы, которые могут исказить обнаружение антител к МкАТ, вызванных его применением. Основываясь на стратегии идентификации и оценки рисков, описанной ниже, стандартную программу изучения иммуногенности в зависимости от уровня идентифицированных рисков допускается сократить (с детальным обоснованием) либо может потребоваться усиление такой программы. Во всех случаях заявитель должен провести тщательную идентификацию рисков, учитывающую свойства препарата и его предлагаемое применение.
Предварительные данные
Необходимо учитывать имеющиеся данные или их отсутствие о прочих аналогичных МкАТ (например, связывающихся с тем же классом мишеней, экспрессирующихся теми же экспрессирующими системами). Если методология обнаружения антител к МкАТ или выявления клинических последствий (например, остаточная концентрация МкАТ, ФД-параметры и эффект от терапии МкАТ) антител к МкАТ недостаточно чувствительна, то восприятие риска может быть завышено. В таких случаях целесообразно осуществлять более тщательное наблюдение за динамикой анти-МкАТ ответа, соотнося его с терапевтическими исходами.
Структура МкАТ
Антитела могут вырабатываться к различным эпитопам, представляющим собой разные части молекулы МкАТ, например, вариабельным или константным участкам. Распознавание гетерологичных (например, последовательностей грызунов или химерных МкАТ) антител в качестве чужеродных служит основной причиной антитело-опосредованного иммунитета, а собственные антитела могут вырабатываться к любой их части. В случае гуманизированных или полностью человеческих последовательностей МкАТ, имеющих аминокислотные последовательности только иммуноглобулина человека, иммунный ответ проявляется формированием, в основном, антиидиотипических антител, специфичных к гипервариабельной последовательности регионов и определяющих комплементарность связывания с антигеном, которые, с большой долей вероятности, могут приводить к снижению клинической эффективности и ответа на терапию МкАТ.
Вместе с тем в некоторых случаях антитела могут вырабатываться к константному участку человеческих и гуманизированных МкАТ, что может влиять на их эффекторные функции и сказываться на клинической эффективности МкАТ. Клинический опыт применения новых конструкций, основанных на МкАТ, ограничен, что также может повышать восприятие риска. Необходимо уделять особое внимание препаратам следующих поколений, например, биспецифичным МкАТ и МкАТ-фрагментам, а также их способности оголять скрытые антигенные детерминанты.
Измененные профили гликозилирования могут снижать или повышать иммуногенные свойства молекулы (например, изменение экранирования белкового остова). Нетипичные профили гликозилирования, например, встречающиеся в начале использования новых экспрессирующих систем, могут представлять повышенный риск иммуногенности по сравнению с широко используемыми экспрессирующими системами.
К другим факторам, влияющим на иммуногенность, относятся производственные примеси и прочие показатели качества. Следовательно, могут потребоваться более глубокие аналитические и клинические подходы, направленные на оценку, установление характеристик и возможное ослабление таких потенциальных рисков, необходимо должным образом идентифицировать риски, обусловленные качеством препарата. Например, МкАТ к мишени, в отношении которой накоплен значительный опыт, но которое производится с помощью новой экспрессирующей системы, может иметь менее воспринимаемый риск с точки зрения его механизма действия, но повышенный риск с точки зрения потенциального влияния примесей вследствие недостаточности сведений об их безопасности.
Механизм действия
Необходимо должным образом установить характеристики и всесторонне изучить механизм действия МкАТ (например, цитолитический, апоптотический) и, особенно, свойства молекулы-мишени (например, угнетение или стимуляция иммунитета). Антитела к МкАТ, мишенью которых является идиотип МкАТ, как правило, снижают эффективность. Необходимо аналогичным образом тщательно изучить влияние антител к МкАТ, распознающих аллотипические или другие участки, поскольку образование иммунных комплексов может привести к нежелательным реакциям у реципиента.
Непрямые эффекты антител, выработанные в ответ на МкАТ, также могут представлять важность, например, МкАТ, мишенью которых являются молекулы, вовлеченные в сигнальные каскады, могут индуцировать антитела, перекрестно связывающиеся с молекулами-мишенями, действуя в качестве агониста, что может привести к повышенной активации иммунной системы и, возможно, вылиться в синдромы высвобождения цитокинов. На уровне отдельного пациента такое достаточно сложно спрогнозировать. В отношении МкАТ-агонистов и МкАТ, перекрестное связывание которых может привести к активации иммунитета, заявители должны предусмотреть тщательное наблюдение за пациентами в ходе ранних клинических исследований на предмет таких явлений.
Клинические факторы
Значительное влияние на иммуногенность оказывают клинические факторы. Иммуногенность к МкАТ может зависеть от возраста, например, метаболизм белков у детей и взрослых различается, что может привести к различиям в иммуногенности, к примеру, антитела, применяемые при ювенильном артрите по сравнению с ревматоидным артритом в сопоставимых дозах. Введение аналогичных (подобных) или родственных антител в анамнезе также может повлиять на иммуногенность. Лекарственные препараты МкАТ, используемые с прерывистой (интермиттирующей) схемой введения (например, с различными интервалами между введениями препарата), могут иметь более высокую вероятность проявления иммуногенности, чем при использовании препаратов в режиме регулярного введения или по цикличным схемам.
Наличие у антител к МкАТ клинически значимых эффектов определяется участком связывания антитела, его аффинностью к МкАТ, а также его титром. Антитела к МкАТ могут быть преходящими и исчезать в ходе лечения или, наоборот, персистировать на протяжении всего лечения и даже дольше. Выработка антител к одним МкАТ не приводит к каким-либо значимым клиническим последствиям, тогда как их выработка к другим может проявляться снижением эффективности или обусловленными лечением нежелательными явлениями.
6.2. Оценка риска
В формирование иммунного ответа на МкАТ вносят вклад множество факторов, требующих учета в ходе оценки рисков.
Факторы, влияющие на частоту и тяжесть иммунного ответа на МкАТ (факторы риска, зависящие от препарата, процесса производства и специфики заболевания и (или) пациентов), могут быть положены в основу подхода, согласно которому эти факторы риска характеризуют с позиции доступности и выполнимости их минимизации в стратегиях оценки (или идентификации) риска.
Идентификация рисков, основанная на обсуждаемых выше факторах, ведет к оценке, объединяющей отдельные клинические риски и надлежащим образом спланированную программу изучения иммуногенности, являющуюся частью клинической разработки. Оценка рисков требует междисциплинарного подхода, учитывающего все выявленные риски, например, обусловленные стратегией контроля качества препарата, включая состав препарата, обоснование пределов приемлемости по родственным вариантам и родственным примесям. Это также подразумевает, что если на различных этапах разработки препарата происходит изменение МкАТ, оценка совокупных рисков должна проводиться при каждом исследовании сопоставимости, проводимом в ходе разработки.
Таким образом, основной стороной оценки рисков является анализ частоты возникновения и клинические последствия нежелательного иммунного ответа, а также возможность предотвращения таких последствий, их правильного определения и (или) медицинской коррекции. В зависимости от выявленных рисков и доступных мер наблюдения и снижения таких рисков, программа изучения иммуногенности может быть меньшей или превосходить ту, что описана в главе 11 настоящих Правил. Заявители должны обосновать и проанализировать принятый подход.
В зависимости от класса и подкласса МкАТ (влияющих на иммунобиологические функции, например, связывание с Fc-рецепторами) или механизма действия, клинические последствия, обусловленные нежелательным иммунным ответом на отдельные препараты МкАТ, могут различаться. Например, МкАТ могут подвергаться нейтрализации антителами, приводящей к снижению эффективности, или вызывать нежелательные явления, например, инфузионные реакции и (или) образование иммунных комплексов. Такие инфузионные реакции могут быть тяжелыми, но их (не являющихся аллергическими реакциями гиперчувствительности) можно снизить с помощью надлежащих клинических мер, например, премедикации. При снижении эффективности наличие других МкАТ или родственных терапевтических белков, являющихся альтернативным методом лечения, также может служить важным фактором стратегии снижения рисков. Общий принцип: при подаче заявлении о регистрации необходимо представить достаточные данные, позволяющие оценить тяжесть, частоту возникновения и идентифицируемость рисков. Затем (при необходимости) такие риски могут быть подвергнуты более глубокому изучению с помощью пострегистрационных исследований и наблюдения.
В качестве отправной точки при оценке и снижении рисков определенную ценность могут представлять следующие факторы:
стратификация рисков, основанная на принципах их выявления, описанная в предыдущем разделе, совмещенная с факторами, обусловленными препаратом, например, выявление внутренних иммуногенных последовательностей, физико-химический профиль, включая агрегаты и прочие родственные и производственные варианты, сведения о разработке состава, например, растворимость при физиологических pH, расположение антигена-мишени и др.;
сведения о функциональных характеристиках метода количественного определения, описанные в настоящей главе, особенно, в какой степени снижается селективность выбранного формата количественного определения МкАТ вследствие остаточной циркуляции препарата;
при неизбежном несовершенстве метода количественного определения: наличие мер, позволяющих дополнить контроль за антителами к МкАТ, например, определение ФД- или ФК-параметров;
наличие методов количественного определения, позволяющих обнаружить ранний иммунный ответ (например, раннее определение связывающих МкАТ, определение IgM для обнаружения раннего иммунного ответа);
восприимчивость популяции пациентов, терапевтический индекс, аутоиммунный статус, одновременное применение иммунодепрессантов и др.;
по сравнению с другими клиническими областями, снижение эффектов в онкологической практике сложнее поддается обнаружению, поскольку прогрессирование опухоли сложно соотнести с выработкой антител. Прогрессирование заболевания и, как следствие, снижение ответа на терапию через некоторое время наблюдается, как правило, почти у всех пациентов, что может затруднить отличие от эффектов, опосредованных иммуногенностью. Вследствие чего, в ходе клинических исследований может потребоваться более интенсивное изучение, чтобы определить, чего ожидать в пострегистрационных условиях, особенно при наличии альтернативных методов лечения;
введение МкАТ на дому и в стационаре: преимуществами введения МкАТ в стационаре является немедленное купирование инфузионных реакций и анафилаксии (при возникновении таковых), однако подкожное введение МкАТ на дому более удобно пациенту. Таким образом, заявителю необходимо сопоставить риск нежелательного иммунного ответа и его последствий с предлагаемым клиническим применением. Например, МкАТ с повышенной частотой реакций после их подкожного введения в меньшей степени подходят для введения на дому;
доступность альтернативных методов терапии или диагностических процедур в случае снижения эффективности или возникновения инфузионных реакций или анафилаксии.
6.3. Мониторинг и снижение рисков
Следуя такому подходу выявления и оценки рисков, заявители должны тщательно спланировать эту концепцию на раннем этапе разработки препарата, затем, по мере получения новых данных, регулярно пересматривать и обновлять ее в ходе процесса разработки и на протяжении всего жизненного цикла препарата. В начале клинической разработки заявители, если того требуют другие факторы, вправе, к примеру, присвоить повышенный риск МкАТ, несмотря на то, что механизм действия per se необязательно предполагает наличие повышенного риска. По результатам крупных клинических исследований может потребоваться пересмотреть величину риска. В ходе регистрации заявители должны тщательно обосновать и проанализировать совокупную концепцию плана и объема исследований иммуногенности, проведенных в ходе программы разработки. Если в отношении лекарственных препаратов указываются, что они обладают благоприятным иммуногенным потенциалом (например, указание в общей характеристике лекарственного препарата), необходимо представить дополнительные данные, обосновывающие такое указание.
В зависимости от результатов оценки рисков в некоторых случаях в ходе клинической разработки могут потребоваться более тщательные и глубокие исследования. Например, если МкАТ содержит нечеловеческие углеводные структуры, такие как галактозо- -1,3-галактоза, в целях недопущения тяжелой анафилаксии до введения препарата пациентам в некоторых случаях необходимо провести анализ на IgE. Другим примером необходимости анализа на IgE является высокая частота аллергических реакций на первое введение препарата в ходе ранней клинической разработки препарата. Несмотря на то, что определение содержания подклассов IgG или иных классов Ig, например, IgA, как правило, не является стандартным требованием изучения иммуногенности МкАТ, такие исследования могут потребоваться, если будут обнаружены определенные риски (например, назальное введение). При этом для установления нейтрализующей способности и преходящего (персистирующего) характера МкАТ, как правило, требуется многократное взятие образцов.
-1,3-галактоза, в целях недопущения тяжелой анафилаксии до введения препарата пациентам в некоторых случаях необходимо провести анализ на IgE. Другим примером необходимости анализа на IgE является высокая частота аллергических реакций на первое введение препарата в ходе ранней клинической разработки препарата. Несмотря на то, что определение содержания подклассов IgG или иных классов Ig, например, IgA, как правило, не является стандартным требованием изучения иммуногенности МкАТ, такие исследования могут потребоваться, если будут обнаружены определенные риски (например, назальное введение). При этом для установления нейтрализующей способности и преходящего (персистирующего) характера МкАТ, как правило, требуется многократное взятие образцов.
В зависимости от степени выявленного относительного риска, частота и сроки отбора проб и их анализ могут варьировать. На поздних этапах разработки допускается снизить частоту отбора проб МкАТ с меньшим риском при условии того, что нежелательные явления или снижение эффективности не отмечались. Тем не менее на протяжении всей программы разработки необходимо на стандартной основе предусмотреть ведение банка проб. Для препаратов МкАТ, риск применения которых более высокий, отбор проб может быть более частым в течение всего периода клинических исследований. В этом случае рекомендуется анализировать пробы в реальном времени. В ходе клинической разработки может потребоваться одновременное, а также в течение периода регулярного введения, определение содержания антител, ФК-, ФД-маркеров, эффективности, безопасности. Это позволяет оценить клиническую значимость выработки антител, а также изменение их эффекта во времени, которое может быть обусловлено повышением их титра и (или) изменением изотипа/созреванием аффинности антител. Антимоноклональные антитела, не обладающие нейтрализующей способностью, могут косвенно влиять на эффективность, связываясь с препаратом МкАТ или изменяя его фармакокинетические свойства. Именно поэтому определение ФК-параметров может содействовать планированию способов определения антимоноклональных антител.
В целях управления рисками можно использовать результаты количественного определения. Например, если согласно заключению выявления и оценки рисков необходимо раннее выявление иммунного ответа и допускается возможность отмены терапии МкАТ, выработка низкоаффинных IgM может служить индикатором раннего иммунного ответа, а определение IgM может способствовать раннему выявлению пациентов, у которых развивается иммунный ответ. Аналогично, обнаружение связывающих антител, не обладающих нейтрализующей способностью, может служить ранним предвестником последующего образования нейтрализующих антител.
Стратегии ослабления рисков могут, к примеру, включать изучение способов ведения пациентов, у которых обнаружен иммунный ответ, например, возможность учащения введения препарата без угрозы его безопасности и др. Однако необходимо учитывать выполнимость таких действий.
При подаче регистрационного досье заявителям рекомендуется представлять объединенную обобщенную стратегию идентификации, описания, мониторинга, минимизации и снижения рисков. Такой основанный на рисках подход должен также учитывать план управления рисками, в котором анализируются способы идентификации рисков по данным из программы разработки, и потенциальные риски, а также отсутствующие сведения, которые необходимо получить на пострегистрационном этапе.
Глава 13. Фармацевтическая разработка биотехнологических
и биологических препаратов
1. Область применения
Несмотря на то, что некоторые вопросы, освещенные в настоящей главе по фармацевтической разработке, имеют отношение к лекарственным препаратам, содержащим активные фармацевтические субстанции, полученные методом химического синтеза, указанные в ней принципы применяются в отношении биологических или биотехнологических лекарственных препаратов.
2. Общие положения
Физико-химические характеристики биологических (биотехнологических) препаратов и препаратов, полученных методом химического синтеза, различаются, в частности биотехнологические и биологические субстанции характеризуются неустойчивостью и сложностью структуры. Подобные различия могут привести к необходимости отдельного рассмотрения фармацевтических и биофармацевтических аспектов в рамках программы исследований и разработки.
В настоящей главе рассматриваются исследования по разработке состава готовой лекарственной формы и предшествующие им исследования, проведение которых следует предусмотреть в процессе разработки подходящей готовой лекарственной формы для активных фармацевтических субстанций биологического или биотехнологического происхождения. Цель таких исследований заключается в разработке стабильного состава лекарственной формы, чтобы при помощи испытаний, свидетельствующих о стабильности, можно было обеспечить биологическую и химическую сохранность активной фармацевтической субстанции для целей:
предлагаемого медицинского применения;
в ходе процесса производства;
при хранении в течение заданного срока годности. Более того, также следует тщательно изучить параметры процесса, которые могут повлиять на показатели постоянства продукта при масштабировании производства.
При проведении фармацевтической разработки биологических (биотехнологических) препаратов в ходе исследований по разработке состава и предшествующих им исследований необходимо тщательно изучить 3 важных взаимосвязанных области разработки, а именно вопросы стабилизации, совместимости и биологической активности. При разработке лекарственного препарата в соответствии с положениями, представленными в настоящей главе, следует учитывать положения других глав настоящих Правил.
3. Частные вопросы
3.1. Установление характеристик
Необходимо провести подробное установление характеристик, используя физико-химические и биофизические методы, соответствующие виду и свойствам активных фармацевтических субстанций, вспомогательных веществ и лекарственных препаратов, включая размер молекулы, заряд и поверхностные свойства. Эти исследования нацелены на описание структурных элементов, отвечающих за биологическую активность, например, активных центров, рецептор- и лиганд-связывающих участков, а также характеристик, отвечающих за передачу сигнала. Они могут также характеризовать соответствующие свойства, потенциально влияющие на фармакокинетику препарата in vivo после его введения, включая сайт-специфичную доставку.
Необходимо также тщательно изучить все физико-химические взаимодействия между активной фармацевтической субстанцией и вспомогательными веществами. В случае с отдельными вирусными и бактериальными вакцинами установление физико-химических и биофизических характеристик может оказаться недостаточным, поэтому в соответствующих случаях необходимо предусмотреть установление биологических характеристик, например иммуногенность.
3.2. Процесс производства
Биологические и биотехнологические препараты отличаются от своих химически синтезированных аналогов процессом производства и его влиянием на качество и безопасность лекарственного препарата. Качество биологических препаратов определяется выбранным процессом и технологией производства. Незначительные изменения процесса производства могут воздействовать на качество лекарственного препарата, поэтому разработка процесса производства имеет первостепенную важность для всех биологических препаратов, включая вакцины, биотехнологические препараты, препараты крови и олигонуклеотидные препараты, такие как ДНК-вакцины и препараты для генной терапии. В рамках стратегии разработки для обеспечения постоянства следует тщательно изучить производственные параметры, способные повлиять на качество и стабильность активной фармацевтической субстанции в составе лекарственной формы, например, pH, нагревание. Если для обеспечения поступления пациенту достаточного количества активной фармацевтической субстанции при введении лекарственного препарата необходим избыток (например, в случае с предварительно заполненным шприцем), следует представить удовлетворительное обоснование с соответствующими подтверждающими данными.
В силу своих физико-химических свойств, как правило, невозможно выполнить терминальную стерилизацию биологических препаратов в готовом контейнере путем автоклавирования. В большинстве случаев стерилизацию проводят перед фасовкой методом мембранной фильтрации. В связи с этим производство препарата в заданных, хорошо контролируемых асептических условиях считается достаточным.
3.3. Избытки
По результатам этих испытаний стабильности в состав определенных биологических (биотехнологических) препаратов, например, вакцин, допускается включать избытки с целью обеспечения сохранения необходимой биологической активности (biological activity) и активности (potency) в течение всего срока годности при заданных условиях хранения. При включении любых излишков необходимо также учитывать вариабельность используемого биологического метода количественного определения активности, особенно если для подобного определения необходимо проведение биологического анализа in vivo.
3.4. Совместимость
Накоплены бесспорные доказательства того, что белки могут химически взаимодействовать со вспомогательными веществами в составе лекарственного препарата, например, с образованием потенциально иммуногенных аддуктов.
В процессе разработки любого препарата необходимо изучить возможные взаимодействия между первичной упаковкой и самим продуктом, чтобы свести к минимуму любое снижение активности (potency) или биологической активности лекарственного препарата, которые могут возникнуть в результате сорбции при хранении. Поскольку белки являются амфифильными полиэлектролитами, они проявляют некоторую степень поверхностной активности. В связи с этим может произойти адсорбционная денатурация за счет диффузии молекул нативного белка к поверхности раздела фаз и их последующей адсорбции, деспирализации полипептидных цепей на поверхности раздела фаз, агрегации поверхностного денатурированного белка с образованием коагулята.
Если лекарственной формой препарата (например, гормона роста) является лиофилизат для приготовления раствора для инъекций, упакованный в двухкамерный контейнер, содержимое которого восстанавливают перед применением, следует обратить особое внимание на обеспечение правильного восстановления лиофилизата или, в случае суспензии, гомогенности ресуспендирования и однородности дозирования. Эти данные должны быть в обязательном порядке отражены в соответствующих разделах листка-вкладыша и общей характеристики лекарственного препарата.
Также следует тщательно изучить совместимость с другими активными фармацевтическими субстанциями, например, в отношении комбинированных вакцин для подкожного, внутримышечного введения или приема внутрь. Речь идет как о живых аттенуированных вакцинах, например, вакцине для профилактики кори, паротита и краснухи, так и об инактивированных вакцинах, например, АКДС и вакцине для профилактики инфекции, вызванной Haemophilus influenzae типа b, когда комбинируемые компоненты изменяют антигенные свойства отдельных элементов.
Необходимо подробно проанализировать клиническую значимость всех полученных нежелательных результатов. Необходимо тщательно изучить влияние адъюванта на достижение достаточного иммунного ответа.
3.5. Стабильность активных фармацевтических субстанций
В ходе исследований, предшествующих исследованиям состава препарата, необходимо определить стабильность активной фармацевтической субстанции. Чтобы белковая молекула сохранила свои биологические свойства, она должна обладать правильной конформацией с целью взаимодействия с рецепторами-мишенями в активном состоянии. Вследствие иерархической организации белковой структуры обычно существует более 1 механизма деградации, как физической, так и химической. В связи с этим, если применимо, с помощью соответствующего множества физико- и биохимических методов необходимо определить пути деградации, включая ее механизмы и кинетику.
Стабильность активной фармацевтической субстанции в препаратах на основе ДНК необходимо оценивать с учетом кинетики деградации в отношении, например, депуринизации и 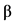 -элиминации.
-элиминации.
Полученные результаты можно использовать с целью минимизации деградации, определив влияние состава препарата, изменений условий процесса производства и хранения, опосредуемых, например, температурой, pH, содержанием солей, механическим стрессом и др. на сохранность действующего вещества.
3.6. Стабильность лекарственного препарата
Сведения о стандартных испытаниях стабильности приведены в главе 8 настоящих Правил.
В связи с неустойчивостью, присущей отдельным биологическим (биотехнологическим) лекарственным препаратам, в состав лекарственной формы или готового препарата (на основании экспериментальных данных) добавляют соответствующее количество подходящих вспомогательных веществ. Это необходимо для придания большей стабильности активной фармацевтической субстанции, например, в отношении ее первичной или третичной структуры, каждая из которых может напрямую влиять на биологическую активность лекарственного препарата, например факторов свертывания крови. Подобные изменения могут возникать в процессе производства и (или) во время хранения.
Необходимо изучить стабильность лекарственной формы или активной фармацевтической субстанции при разных режимах производственного процесса, например, при лиофилизации, в целях оптимизации состава, например, количества лиопротектора, необходимого для обеспечения сохранности активной фармацевтической субстанции.
Вспомогательные вещества и (или) реагенты, используемые в процессе производства активных фармацевтических субстанций или при разработке состава препаратов, могут иметь человеческое или животное происхождение. При фармацевтической разработке неустойчивых биологических препаратов, таких как живые аттенуированные вакцины, желательно разработать и оценить альтернативу использованию материалов человеческого или животного происхождения в процессе производства и при разработке состава. По возможности следует изучать возможность замены альбумина человека как вспомогательного вещества.
Глава 14. Требования к документации по управлению
качеством биологических препаратов, исследуемых
в клинических исследованиях
1. Область применения
Уполномоченными органами государств-членов устанавливаются требования к получению заявителями разрешения на проведение клинического исследования лекарственного препарата для медицинского применения, уведомлению о существенных поправках и заявлению о завершении исследования, а также требования к данным в досье исследуемого лекарственного препарата (далее - ДИЛП), представляемым для получения разрешения на проведение клинического исследования.
В настоящей главе разъясняются частные требования к документации по биологическому, химическому и фармацевтическому качеству исследуемых лекарственных препаратов, содержащих биологические (биотехнологические) активные фармацевтические субстанции.
Кроме того, в настоящей главе содержатся примеры изменений документации в отношении биологического, химического и фармацевтического качества исследуемых лекарственных препаратов, рассматриваемые, как правило, в качестве "существенных".
Требования настоящей главы применимы к белкам и полипептидам, их производным и препаратам, компонентами которых они являются (например, к конъюгатам). Эти белки и полипептиды получают из рекомбинантных или нерекомбинантных экспрессирующих систем клеточных культур, они хорошо поддаются очистке и установлению свойств с помощью соответствующего набора аналитических методик.
Эти принципы могут также применяться к другим типам препаратов, например белкам и полипептидам, выделенным из тканей и жидкостей организма.
2. Данные о биологическом, химическом и фармацевтическом
качестве биологических лекарственных препаратов,
исследуемых в клинических исследованиях
S. Активная фармацевтическая субстанция
Ссылки на мастер-файл на активную фармацевтическую субстанцию и сертификат соответствия Европейского директората по качеству лекарственных средств (CEP) в отношении биологических (биотехнологических) активных фармацевтических субстанций не применимы и не допускаются.
S.1. Общая информация
S.1.1. Номенклатура.
Необходимо представить информацию по номенклатуре активной фармацевтической субстанции (например, предлагаемое международное непатентованное наименование (МНН), фармакопейное наименование, патентованное наименование (торговое наименование), код компании, другие наименования или коды при наличии).
S.1.2. Структура.
Необходимо представить краткое описание предполагаемой структуры, включая данные о структурах высокого порядка, схему аминокислотной последовательности с указанием участков гликозилирования и других посттрансляционных модификаций, а также привести значение относительной молекулярной массы.
S.1.3. Общие свойства.
Необходимо представить перечень физико-химических и других важных свойств активной фармацевтической субстанции, включая биологическую активность (особую способность или свойство препарата оказывать определенный биологический эффект). Необходимо проанализировать предлагаемый механизм действия.
S.2. Процесс производства активной
фармацевтической субстанции
S.2.1. Производитель (производители).
Необходимо представить название, адрес и ответственность каждого производителя, включая контрактных производителей, а также каждой предлагаемой производственной площадки, вовлеченной в производство и контроль качества.
S.2.2. Описание процесса производства и его контроля.
Необходимо должным образом описать производственный процесс и контроль процесса производства. Обычно производственный процесс начинается с контейнера банка клеток и включает культивирование клеток, сбор (сборы), очистку, модификацию продукта и его фасовку. Следует также описать условия хранения и транспортировки.
Необходимо представить схему, отражающую последовательность всех стадий производства, включая внутрипроизводственные испытания. Результаты внутрипроизводственных испытаний допускается выражать в виде пределов действия или предварительных критериев приемлемости. По мере накопления данных в ходе разработки необходимо представить более подробные результаты внутрипроизводственных испытаний и критерии. Критерии приемлемости подлежат пересмотру.
Необходимо определить размер серии и масштаб производства, включая информацию о любом объединении сборов или промежуточных продуктов.
Необходимо описать и обосновать процедуры повторной обработки в ходе процесса производства активной фармацевтической субстанции (например, невыдерживание испытания на целостность фильтра).
S.2.3. Контроль качества материалов.
Сырье и исходные материалы. Необходимо представить перечень материалов, использованных в производстве фармацевтической субстанции (например, сырье, исходные материалы, питательные среды, факторы роста, хроматографические смолы, растворители, реактивы), с указанием на каком этапе процесса используется каждый из них. Необходимо представить ссылки на стандарты качества (например, фармакопейные статьи или собственные спецификации производителя). Необходимо представить сведения о качестве и контроле качества нефармакопейных материалов. Необходимо представить сведения, подтверждающие, что материалы (включая материалы из биологических источников, например, компоненты среды, моноклональные антитела, ферменты) удовлетворяют стандартам их целевого назначения.
В отношении всего сырья биологического происхождения (включая использованные при формировании банка клеток) необходимо указать источник и соответствующий этап процесса производства, в котором оно использовалось. В приложении A.2 модуля 3 регистрационного досье необходимо представить резюме о вирусной безопасности материалов биологического происхождения (регистрационное досье формируется до подачи заявления на регистрацию лекарственного препарата).
Источник, история и образование клеточного субстрата. Необходимо представить резюме об источнике и наработке (блок-схема последовательности этапов) клеточного субстрата, анализ экспрессирующего вектора, использованного для генетической модификации клеток и включенного в исходную клетку (клетку-хозяина) для создания главного банка клеток и стратегии усиления и контроля производства соответствующего гена (согласно принципам главы 1 настоящих Правил).
Система банка клеток, установление характеристик и испытания. Главный банк клеток необходимо создать до начала I фазы исследований. Не во всех случаях требуется формировать рабочий банк клеток (РБК).
Необходимо представить сведения о получении, квалификации и формировании банков клеток. Необходимо описать ГБК и (или) РБК и представить результаты проведенных испытаний. Получение и установление характеристик банков клеток необходимо осуществлять в соответствии с принципами главы 1 настоящих Правил.
Клеточные банки необходимо описывать с помощью соответствующих фенотипических или генотипических маркеров, чтобы обеспечить подлинность, жизнеспособность и чистоту клеток, использованных в производстве.
До начала клинических исследований необходимо подтвердить последовательность нуклеиновой кислоты экспрессирующей кассеты, включая последовательность кодирующего участка.
При необходимости в приложении A.2 модуля 3 регистрационного досье необходимо представить анализ безопасности относительно посторонних агентов и квалификации банков клеток, использованных для производства активной фармацевтической субстанции.
Стабильность клеточного субстрата. Необходимо представить все доступные данные о стабильности клеточного субстрата.
S.2.4. Контроль критических этапов и промежуточных продуктов.
Необходимо представить описание испытаний и критерии приемлемости для контроля критических этапов процесса производства. Вследствие ограниченности данных на ранних этапах разработки (I - II фазы) полные сведения могут отсутствовать.
Необходимо, используя дополнительные данные, обосновать сроки и условия хранения промежуточных продуктов.
S.2.5. Валидация и (или) оценка процесса.
На протяжении всего хода разработки необходимо собирать данные о валидации (оценке) процесса производства, однако такие данные для получения разрешения на проведение клинического исследования не требуются.
В приложении A.2 модуля 3 регистрационного досье необходимо представить сведения об этапах производства, направленных на элиминацию и инактивацию вирусных контаминантов.
S.2.6. Разработка процесса производства.
Совершенствование процесса производства. Процессы производства и стратегии их контроля подлежат непрерывному совершенствованию и оптимизации, особенно на этапе разработки и на ранних фазах клинических исследований. Такое совершенствование и оптимизация рассматриваются в качестве естественного хода разработки, что требует соответствующего описания в документах, представляемых для получения разрешения на проведение клинического исследования. Необходимо обобщить сведения об изменениях процесса производства и его контроля и представить обоснование таких изменений. В целях определения связи между сериями до внесения и после внесения изменений в описании необходимо однозначно представить сведения, позволяющие идентифицировать варианты процессов производства, использованных для получения каждой серии, использованной в доклинических и клинических исследованиях. Для описания эволюции процесса производства допускается представлять сравнительные блок-схемы и (или) перечень изменений процесса производства. Изменения процесса производства могут потребовать адаптации внутрипроизводственных и выпускающих испытаний, в связи с чем после внесения изменений, такие испытания и соответствующие критерии приемлемости требуют пересмотра.
Исследования сопоставимости. В зависимости от последствий внесения изменений и этапа разработки в целях обеспечения отсутствия нежелательного влияния на клинические характеристики лекарственного препарата могут потребоваться исследования сопоставимости. Основная цель таких исследований - обеспечение пригодности препарата после его изменения для клинических исследований и отсутствия угрозы для безопасности пациентов, включенных в клинические исследования.
Исследования сопоставимости необходимо проводить поэтапно, включая сравнение параметров качества активной фармацевтической субстанции и значимых промежуточных продуктов, используя подходящие аналитические методы. К аналитическим методам, как правило, относятся стандартные испытания, которые при необходимости допускается дополнять вспомогательными испытаниями для установления характеристик (включая ортогональные методы). Если накопленного производителем опыта и прочих значимых сведений для оценки рисков, обусловленных изменениями, недостаточно или ожидаются потенциальные риски для пациентов, исследований сопоставимости, основанных исключительно на изучении вопросов качества, может быть недостаточно.
В ходе ранних фаз доклинических и клинических исследований исследования сопоставимости, как правило, не столь многочисленны, как для зарегистрированного лекарственного препарата. В клинических исследованиях, впервые проводимых у человека, рекомендуется применять исследуемый лекарственный препарат, отражающий свойства материала, использованного в доклинических исследованиях.
S.3. Описание характеристик активной
фармацевтической субстанции
S.3.1. Установление структуры и других характеристик.
В целях составления спецификации необходимо с помощью соответствующих методик охарактеризовать биотехнологическую или биологическую фармацевтическую субстанцию (включая определение физико-химических свойств, биологической активности, иммунохимических свойств, чистоты и примесей). Не допускается ограничиваться ссылками на литературные данные. На этапе разработки до начала I фазы клинических исследований и при необходимости после значительных изменений процесса производства необходимо провести надлежащее установление характеристик.
В отношении целевого продукта необходимо представить подробные сведения о первичной, вторичной структуре и структуре более высокого порядка, посттрансляционных (например, гликоформах) и прочих модификациях.
Необходимо представить подробные сведения о биологической активности (особой способности или свойстве препарата оказывать определенный биологический эффект). До начала исследований фазы I, как правило, необходимо, используя соответствующие надежные и квалифицированные методы, определить биологическую активность. Степень установления характеристик на более поздних фазах увеличится.
Необходимо представить научное обоснование использованных методов установления характеристик и обоснование их пригодности.
S.3.2. Примеси.
Необходимо изучить производственные (например, белки клетки-хозяина, ДНК клетки-хозяина, остатки питательных сред, экстрагируемые из колонок вещества) и родственные примеси (например, прекурсоры, расщепленные формы, продукты деградации, агрегаты). Необходимо представить количественные данные о примесях, включая максимальное содержание в высшей клинической дозе. Может потребоваться оценка очистки от определенных производственных примесей (например, пеногасителей).
В отношении некоторых примесей необходимо обосновать представление исключительно качественных (без количественных) данных о них.
S.4. Контроль качества активной фармацевтической субстанции
В ходе фаз клинических исследований, если данные по валидации неполные, в целях подтверждения фармацевтического качества, постоянства и сопоставимости фармацевтической субстанции после изменения процесса производства необходимо знать показатели качества для контроля фармацевтической субстанции. Вследствие чего показатели качества, контролируемые на протяжении всего процесса разработки, не должны ограничиваться испытаниями, включенными в спецификацию, для которых установлены предварительные критерии приемлемости.
S.4.1. Спецификация.
В спецификации серий активной фармацевтической субстанции, которые будут использоваться в клинических исследованиях, необходимо описать их критерии приемлемости, а также испытания, использованные для установления надлежащего контроля за качеством активной фармацевтической субстанции. Испытания на количественное содержание, подлинность и примеси являются обязательными. В отсутствие должного обоснования необходимо включить испытание на биологическую активность. Необходимо установить верхние пределы содержания примесей, учитывая показатели безопасности. Необходимо охарактеризовать микробиологическую чистоту активной фармацевтической субстанции.
Поскольку критерии приемлемости, как правило, основаны на ограниченном количестве серий, использованных в разработке, и серий, использованных в доклинических и клинических исследованиях, они изначально являются предварительными и требуют доработки и коррекции в ходе последующей разработки.
Необходимо также документировать свойства продукта, которые на определенном этапе разработки полностью не охарактеризованы, или в отношении которых имеются лишь ограниченные данные для установления критериев приемлемости. Как следствие, такие свойства продукта допускается включать в спецификацию без заранее установленных пределов приемлемости. Результаты необходимо представить в разделе по посерийному анализу (в соответствии с разделом S.4.4 настоящих Правил).
Дополнительная информация
по клиническим исследованиям фазы II и III
По мере накопления информации и опыта может потребоваться добавление или исключение параметров и модификация аналитических методик. Спецификации и критерии приемлемости, установленные для предыдущих испытаний, следует пересмотреть и при необходимости скорректировать в соответствии с текущей стадией разработки.
S.4.2. Аналитические методики.
Необходимо указать аналитические методики для всех испытаний фармацевтической субстанции, включенных в спецификацию (например, хроматографические методы, биологические методы и др.), включая испытания без пределов приемлемости. Необходимо представить краткое описание всех нефармакопейных аналитических методик (способ проведения анализа).
В отношении методов, соответствующих требованиям статей Фармакопеи Союза, фармакопеи государств-членов, ведущих зарубежных фармакопей в соответствии с концепцией гармонизации, допускается ссылка на соответствующую статью.
S.4.3. Валидация аналитических методик.
Валидация аналитических методик в ходе клинической разработки рассматривается в качестве постепенного процесса.
Аналитические методики, описанные в общей главе Фармакопеи Союза, фармакопеи государств-членов, ведущих зарубежных фармакопеях в соответствии с концепцией гармонизации или связанные с частной фармакопейной статьей, обычно считаются валидированными.
Необходимо подтвердить пригодность использованных в клинических исследованиях фазы I аналитических методов. Пределы приемлемости (например, пределы приемлемости для определения содержания примесей) и параметры (специфичность, линейность, аналитическая область, правильность, прецизионность, пределы количественного определения и обнаружения) проведения валидации аналитических методик необходимо представить в табличной форме.
Информация по клиническим исследованиям фазы II и фазы III. Необходимо подтвердить пригодность использованных аналитических методик. Необходимо представить в форме таблицы резюме результатов проведенной валидации (например, результаты или значения специфичности, линейности, аналитической области, правильности, прецизионности, пределов количественного определения и обнаружения). Полный отчет о валидации представлять не требуется.
S.4.4. Посерийный анализ.
Поскольку первоначально спецификация может быть очень широкой, для оценки качества важны фактические данные по сериям. Для количественных параметров необходимо представить фактические численные значения.
Задача настоящего раздела - подтвердить качество серий (соответствие предварительной спецификации), подлежащих применению в конкретном клиническом исследовании. Для клинических исследований ранней фазы, для которых обычно характерно ограниченное число серий, необходимо представить результаты по релевантным доклиническим и клиническим сериям, включая результаты анализа серий, подлежащих применению в данном клиническом исследовании. Однако при более продолжительном производстве при должном обосновании допускается представить только результаты нескольких репрезентативных серий.
Наряду с использованием серий необходимо представить сведения о номере серии, ее размере, производственной площадке, дате производства, методах контроля, критериях приемлемости и результатах испытаний. Необходимо описать процесс производства каждой серии.
S.4.5. Обоснование спецификации.
Необходимо представить подробное обоснование всех показателей качества, включенных в спецификацию, и критериев приемлемости по чистоте, примесям, биологической активности и другим показателям качества, которые могут повлиять на функциональные свойства лекарственного препарата. При обосновании необходимо руководствоваться соответствующими данными по разработке, данными о сериях, использованных в доклинических и (или) клинических исследованиях, и результатами исследований стабильности, учитывая методы, использованные для их контроля. Считается, что в ходе ранней клинической разработки критерии приемлемости могут быть шире и могут не отражать возможности технологического процесса. Если опыт ограничен, в фазы I - II допускается устанавливать более широкие пределы. Однако для тех показателей качества, которые могут повлиять на безопасность пациента, необходимо тщательно обосновывать пределы, принимая во внимание доступную информацию (например, возможности технологического процесса, вид лекарственного препарата, дозу, продолжительность введения и др.). Необходимо обосновать значимость выбранной методики количественного определения биологической активности и предлагаемых пределов ее приемлемости.
Необходимо описать и обосновать изменения, вносимые в ранее использовавшуюся спецификацию (например, включение или исключение параметров, расширение критериев приемлемости).
S.5. Стандартные образцы или материалы
Учитывая свойства биологических (биотехнологических) препаратов, в целях обеспечения постоянства между различными сериями исследуемого лекарственного препарата, а также сопоставимости препарата, подлежащего коммерческой реализации, с препаратом, использованным в клинических исследованиях, а также описания связи между процессом разработки и промышленным производством необходимо использовать надлежащим образом охарактеризованный стандартный материал. Установление характеристик стандартного материала необходимо осуществлять с помощью современных аналитических методов, которые необходимо описать надлежащим образом. Необходимо представить сведения о процессе производства, использованного для получения стандартного материала.
Если в ходе клинической разработки использовался не 1 стандартный образец, необходимо представить историю квалификации, описывающую поддержание связи между различными стандартами.
В качестве первичного стандарта необходимо использовать международные или фармакопейные стандартные образцы (при наличии). Однако следует учитывать, что использование международного стандартного образца или фармакопейного стандартного образца может быть ограничено определенными методами испытаний, например, на биологическую активность. Если международный стандартный образец или фармакопейный стандартный образец отсутствует, необходимо произвести собственный стандартный материал.
S.6. Система упаковки (укупорки)
Необходимо указать упаковочный материал, используемый для активной фармацевтической субстанции. Следует принять во внимание возможность взаимодействия между активной фармацевтической субстанцией и первичной (внутренней) упаковкой.
S.7. Стабильность
Резюме и выводы по стабильности
(протокол (материал) и метод)
Необходимо представить программу изучения стабильности, охватывающую предлагаемый срок хранения активной фармацевтической субстанции, включая спецификацию, аналитические методики и интервалы между испытаниями. Интервалы между испытаниями должны соответствовать главе 8 настоящих Правил.
Качество серий активной фармацевтической субстанции, включенных в программу изучения стабильности, должно отражать качество материала, подлежащего использованию в планируемом клиническом исследовании.
Активную фармацевтическую субстанцию, включенную в программу изучения стабильности, необходимо хранить в контейнерах, в которых используются те же вид и материалы системы упаковки (укупорки), которые будут использоваться в отношении активной фармацевтической субстанции, подлежащей использованию в клиническом исследовании. При изучении стабильности активной фармацевтической субстанции допускается использовать контейнеры меньшего размера.
В исследованиях необходимо изучить стабильность активной фармацевтической субстанции в предлагаемых условиях хранения. Рекомендуется проводить исследования ускоренного хранения и стресс-стабильности, поскольку они позволяют понять профиль деградации и служат в качестве подкрепляющих данных при увеличении срока годности.
Чтобы удостовериться в том, что будут выявлены изменения профиля чистоты (примесей) и биологической активности активной фармацевтической субстанции, в программу изучения стабильности необходимо включить методы-индикаторы стабильности. В отсутствие должного обоснования в программу необходимо включить методику количественного определения активности.
Описанный в документе Союза, посвященном изучению стабильности, период повторного испытания для биологических (биотехнологических) активных фармацевтических субстанций не применим.
Данные (результаты) исследования стабильности
Необходимо представить данные по стабильности по меньшей мере одной серии, отражающей процесс производства материала, который будет использоваться в клиническом исследовании. Кроме того, допускается представить данные по стабильности соответствующих серий, использовавшихся в разработке, или серий, произведенных с помощью предыдущего процесса производства. Такие данные допускается использовать для определения срока годности активной фармацевтической субстанции при условии представления обоснования того, что качество серии отражает качество материала, подлежащего изучению в клинических исследованиях.
Все значимые данные по стабильности необходимо обобщить в таблице с указанием испытанных серий, даты их производства, версии процесса производства, состава, условий хранения, временных точек, аналитических методик, критериев приемлемости и результатов.
Необходимо представить фактические числовые значения количественных параметров. Необходимо проанализировать все выявленные тенденции.
Для учета объема доступных данных и нарастающих знаний о стабильности активной фармацевтической субстанции в ходе различных фаз клинической разработки к стабильности необходимо предъявлять более строгие требования. К фазе III заявитель должен всесторонне понимать профиль стабильности активной фармацевтической субстанции.
Установление срока годности
Необходимо описать заявленный срок годности активной фармацевтической субстанции в предлагаемых условиях хранения, сопроводив описание анализом доступных данных. Необходимо проанализировать все выявленные тенденции.
Как описано в главе 8 настоящих Правил, запрашиваемый период хранения необходимо определять на основании результатов исследований естественного хранения в реальном времени и при реальной температуре. Допускается увеличение срока годности сверх данных по стабильности в реальном времени, при условии обоснования соответствующими данными, в том числе результатами исследований ускоренного хранения.
Максимальный срок годности после такого увеличения допускается увеличивать не более чем в 2 раза и не более чем на 12 месяцев по сравнению с данными по стабильности, полученными на репрезентативных сериях. Однако увеличение срока годности, превышающее предполагаемую длительность исследований естественного хранения, не допускается.
При составлении программы изучения стабильности допускается опираться на предыдущие данные, включая платформенные технологии. Однако сами по себе такие данные не являются достаточными для обоснования срока годности исследуемого лекарственного препарата.
При планировании увеличения срока годности заявитель должен принять обязательство выполнить предложенную программу стабильности в соответствии с представленным протоколом и при возникновении непредвиденных ситуаций уведомить о них уполномоченные органы, а также предложить план корректирующих мероприятий.
Сведения об увеличении срока годности путем внесения существенной поправки представлены в подразделе 4 настоящей главы.
P. Исследуемый лекарственный препарат
P.1. Описание и состав исследуемого
лекарственного препарата
Следует указать качественный и количественный состав исследуемого лекарственного препарата. Предоставляемая информация должна включать:
краткое или табличное описание лекарственной формы;
состав, то есть перечень всех компонентов лекарственной формы и их содержание в расчете на единицу (включая избытки, при наличии), функциональное назначение компонентов, ссылки на их стандарты качества (например, на фармакопейные статьи или спецификации производителей);
описание прилагающихся разбавителей;
краткое описание типа контейнера и системы укупорки, используемых для лекарственной формы и прилагающегося разбавителя для ее приготовления, если это применимо.
P.2. Фармацевтическая разработка
На ранних этапах разработки информации для включения в настоящий раздел может быть недостаточно.
Необходимо представить краткое описание разработки состава, включая обоснование новой лекарственной формы или нового вспомогательного вещества.
В отношении лекарственных препаратов, требующих дополнительного приготовления (например, восстановления, разведения, смешивания), необходимо подтвердить совместимость с такими материалами (например, растворителями, разбавителями, средой) и резюмировать метод приготовления (допускается сослаться на полное описание, содержащееся в протоколе клинического исследования).
Необходимо представить обоснование, что комбинирование лекарственной формы и упаковочного материала не нарушает правильное дозирование, например, подтвердить, что препарат не абсорбируется на стенках контейнера или инфузионной системы. Выполнение этого требования особенно важно для форм выпуска с низкой дозой или высоким разведением. По возможности в клинических исследованиях, впервые проводимых у человека, необходимо удостовериться, что очень низкие дозы вводятся правильно.
Разработка процесса производства
Необходимо описать изменения процесса производства, включая изменения состава и лекарственной формы по сравнению с ранее проведенными клиническими исследованиями. Значительные изменения, например, изменения состава, необходимо обосновать соответствующими исследованиями сопоставимости. В этой связи необходимо следовать положениям, описанным в разделе S.2.6 настоящих Правил. Эти данные необходимо подробно детализировать, чтобы надлежащим образом уяснить суть изменений и оценить возможные последствия для безопасности пациента.
Все изменения состава в ходе клинической разработки необходимо документировать и обосновать с точки зрения их влияния на качество, безопасность, клинические свойства, дозирование и стабильность лекарственного препарата.
P.3. Процесс производства исследуемого
лекарственного препарата
P.3.1. Производитель (производители).
Необходимо представить название, адрес и ответственность каждого производителя, включая контрактных производителей, а также каждой предлагаемой производственной площадки, вовлеченной в производство, испытания и выпуск серии. Если в производстве исследуемого лекарственного препарата участвуют несколько производителей, необходимо четко описать обязанности каждого из них.
P.3.2. Материальный баланс (состав на серию).
Необходимо представить материальный баланс серии (серий), планируемых к использованию в клинических исследованиях. Эти данные должны включать перечень всех компонентов, которые будут использованы. Следует указать размеры или диапазон объема серий.
P.3.3. Описание процесса производства и его контроля.
Необходимо представить схему всех последовательных этапов процесса, включая внутрипроизводственные испытания. Результаты внутрипроизводственных испытаний можно установить в качестве пределов действия или учитывать как предварительные критерии приемлемости. По мере накопления в ходе разработки данных о процессе необходимо представить более подробные результаты внутрипроизводственных испытаний и критериев. Критерии приемлемости подлежат пересмотру.
Большинство препаратов, содержащих рекомбинантные белки и моноклональные антитела, производятся с помощью асептических процессов, рассматриваемых в качестве нестандартных. Нестандартные процессы производства, новые технологии и новые упаковочные процессы требуют подробного описания.
P.3.4. Контроль критических этапов и промежуточных продуктов.
Необходимо описать испытания и критерии приемлемости контроля ключевых этапов процесса производства. Вследствие ограниченности данных на ранних этапах разработки (фаза I и фаза II) полная информация может отсутствовать.
Если для обработки промежуточных продуктов предусматривается их хранение, необходимо описать сроки и условия хранения и обосновать их данными изучения физико-химических, биологических и микробиологических свойств.
Для стерилизации методом фильтрации в досье для получения разрешения на проведение клинического исследования следует указать максимальную приемлемую биологическую нагрузку до фильтрации. В большинстве случаев приемлем показатель в 10 КОЕ/100 мл, в зависимости от соотношения фильтруемого объема и диаметра фильтра. Если это требование не выполняется, необходимо использовать предварительную фильтрацию через антибактериальный фильтр. Таким образом, можно получить желаемую низкую биологическую нагрузку. Вследствие небольших количеств приготовленного лекарственного препарата при достаточном обосновании допускается испытывать 100 мл нефильтрованного (фильтрованного) препарата.
Повторные манипуляции допускаются на конкретных этапах производства (например, повторная фильтрация), только если такие этапы должным образом описаны и обоснованы.
P.3.5. Валидация и (или) оценка процесса.
Если применимо, необходимо кратко описать валидацию асептической обработки и лиофилизацию. В соответствии с приложением N 13 к Правилам надлежащей производственной практики Союза, утверждаемым Комиссией, стандарт валидации процессов стерилизации не должен отличаться от стандартов, предусмотренных для зарегистрированного лекарственного препарата. В досье, в частности, необходимо включить сведения, напрямую затрагивающие безопасность лекарственного препарата (о бионагрузке и циклах добавления среды).
P.4. Контроль качества вспомогательных веществ
P.4.1. Спецификация.
Допустимо использовать ссылки на Фармакопею Союза, национальные фармакопеи государств-членов, ведущие зарубежные фармакопеи в соответствии с Концепцией гармонизации фармакопей государств-членов, утверждаемой Комиссией. По вспомогательным веществам, не включенным в вышеупомянутые стандарты, необходимо представить внутренние спецификации производителя.
P.4.2. Аналитические методики.
При невозможности сослаться на фармакопейные статьи, перечисленные в разделе P.4.1 настоящих Правил, необходимо описать использованные аналитические методики.
P.4.3. Валидация аналитических методик.
Не применимо.
P.4.4. Обоснование спецификаций.
Для нефармакопейных вспомогательных веществ следует обосновать внутренние спецификации производителя, приведенные в разделе P.4.1 настоящих Правил.
P.4.5. Вспомогательные вещества человеческого или животного происхождения.
В отношении вспомогательных веществ человеческого или животного происхождения в приложении A.2 модуля 3 регистрационного досье необходимо представить сведения об оценке безопасности относительно посторонних агентов (например, источники, спецификации, описание проведенных испытаний) и данные по вирусной безопасности в соответствие с главой 3 настоящих Правил. Кроме того, в приложении A.2 модуля 3 регистрационного досье необходимо описать соответствие главе по минимизации риска трансмиссивной губчатой энцефалопатии при применении лекарственных препаратов для медицинского применения в его текущей редакции.
Если в качестве вспомогательного вещества используется альбумин человека или иное вещество, полученное из плазмы, необходимо представить сведения об оценке безопасности относительно посторонних агентов согласно соответствующим главам о лекарственных препаратах, полученных из плазмы. Если полученный из плазмы компонент ранее использовался в зарегистрированном лекарственном препарате, допускается ссылка на это.
P.4.6. Новые вспомогательные вещества.
Для вспомогательного вещества, использованного в лекарственном препарате впервые или при новом пути введения, необходимо представить полное описание процесса производства, установления характеристик и контроля качества со ссылками на дополнительные данные по безопасности (доклинические и (или) клинические) согласно формату активной фармацевтической субстанции.
P.5. Контроль качества исследуемого
лекарственного препарата
P.5.1. Спецификации.
В отношении лекарственного препарата необходимо руководствоваться теми же принципами, которые описаны для составления активной фармацевтической субстанции. В спецификации серий исследуемого лекарственного препарата, которые будут использоваться в клинических исследованиях, необходимо описать испытания и критерии их приемлемости, использованные для установления надлежащего контроля за его качеством. Испытания на количественное содержание, подлинность и примеси являются обязательными. В отношении стерильных лекарственных препаратов обязательно проведение испытаний на стерильность и эндотоксины. При отсутствии должного обоснования необходимо включить испытание на биологическую активность. Необходимо установить верхние пределы содержания примесей, учитывая показатели их безопасности. Они могут потребовать пересмотра и коррекции в ходе последующей разработки.
Критерии приемлемости показателей качества лекарственного препарата требуют учета вопросов безопасности и этапа разработки. Поскольку критерии приемлемости, как правило, основаны на ограниченном количестве серий, использовавшихся в разработке, и серий, использованных в доклинических и клинических исследованиях, они изначально являются предварительными и могут потребовать пересмотра и коррекции в ходе последующей разработки.
Аналитические методики и пределы содержания и биологической активности должны обеспечивать правильное дозирование.
Необходимо установить верхние пределы содержания примесей, не включенных в спецификацию активной фармацевтической субстанции, принимая во внимание вопросы безопасности.
Дополнительная информация по клиническим исследованиям
фазы II и фазы III
По мере накопления данных и опыта может потребоваться дополнение или исключение параметров и модификация аналитических методик. Спецификацию и критерии приемлемости, установленные для ранее проведенных испытаний, следует пересмотреть для клинических исследований фазы III и при необходимости адаптировать к текущим этапом разработки.
P.5.2. Аналитические методики.
Необходимо описать аналитические методики всех испытаний, включенных в спецификацию. В отношении некоторых белков и комплексов, а также инновационных лекарственных форм, может потребоваться большая детализация.
Иные сведения указываются в соответствии с разделом S.4.2.
P.5.3. Валидация аналитических методик.
Указываются сведения в соответствии с разделом S.4.3.
P.5.4. Посерийный анализ.
Поскольку изначально спецификация может быть широкой, для оценки качества необходимо представить фактические данные о сериях. Необходимо представить фактические числовые значения количественных параметров.
Цель настоящего раздела - подтвердить качество серий (соответствие разработанной предварительной спецификации), подлежащих применению в данном клиническом исследовании. В отношении клинических исследований ранней фазы, для которых характерно небольшое количество серий, необходимо представить результаты анализа серий, участвовавших в доклинических и клинических исследованиях, включая результаты анализа серий, подлежащих изучению в данном клиническом исследовании. Однако по мере накопления опыта производства при должном обосновании допускается представить результаты анализа лишь определенного количества репрезентативных серий.
Наряду с использованием серий необходимо представить сведения о номере серии, ее размере, производственной площадке, дате производства, методах контроля, критериях приемлемости и результатах испытаний. Необходимо описать процесс производства каждой серий.
P.5.5. Установление свойств примесей.
Дополнительные примеси и продукты деградации, обнаруженные в исследуемом лекарственном препарате, но не описанные в разделе S.3.2, необходимо соответствующим образом идентифицировать и квалифицировать.
P.5.6. Обоснование спецификации.
При обосновании показателей качества, включенных в спецификацию лекарственного препарата, необходимо руководствоваться спецификацией активной фармацевтической субстанции. Необходимо учесть показатели качества, отражающие стабильность. Необходимо обосновать предложенные критерии приемлемости.
P.6. Стандартные образцы или материалы
В соответствующих случаях необходимо представить параметры установления характеристик стандартного образца.
При необходимости допускается сослаться на раздел S.5.
P.7. Система "контейнер - укупорка"
Необходимо описать предполагаемую первичную упаковку исследуемого лекарственного препарата, подлежащего изучению в клиническом исследовании. Если применимо, следует сослаться на соответствующую фармакопейную статью. Если препарат упакован в нестандартное устройство введения или использованы нефармакопейные материалы, необходимо представить их описание и спецификации. Если применимо, необходимо подтвердить знак Союза о регистрации изделия в качестве вспомогательного медицинского изделия.
В отношении парентеральных лекарственных препаратов, способных взаимодействовать с системой "контейнер - укупорка", могут потребоваться дополнительные данные.
P.8. Стабильность лекарственного препарата
К лекарственному препарату применяются те же требования, что и к активной фармацевтической субстанции, включая программу изучения стабильности, результаты изучения стабильности, определение срока годности (в том числе увеличение срока годности свыше периода, охваченного данными естественного хранения), обязательства по стабильности и пострегистрационное расширение. Результаты исследований стабильности должны подтвердить, что исследуемый лекарственный препарат остается стабильным на протяжении всего периода хранения. Представленные данные должны подтверждать предлагаемый срок годности лекарственного препарата с момента его выпуска до введения пациентам. В протоколе изучения стабильности исследуемого лекарственного препарата необходимо учесть данные, приобретенные по результатам изучения стабильности активной фармацевтической субстанции.
При достаточном обосновании приемлемыми являются исследование крайних вариантов и матричный метод.
В отношении препаратов, предназначенных для применения после восстановления, разведения или смешивания, необходимо представить данные по стабильности готового к применению лекарственного препарата. Такие данные не требуются, если приготовленный лекарственный препарат после вскрытия упаковки или восстановления подлежит немедленному применению.
3. Приложения
A.1. Производственные помещения и оборудование
Не применимо.
A.2. Оценка безопасности относительно посторонних агентов
Необходимо идентифицировать все материалы человеческого или животного происхождения, использованные в процессе производства как активной фармацевтической субстанции, так и лекарственного препарата, и материалы, соприкасающиеся с активной фармацевтической субстанцией или лекарственным препаратом в ходе процесса производства. В настоящем разделе необходимо представить данные по оценке риска с точки зрения потенциальной контаминации посторонними агентами.
Агенты трансмиссивной губчатой энцефалопатии
Необходимо представить подробные сведения по недопущению и контролю агентов трансмиссивной губчатой энцефалопатии. Такие сведения, к примеру, могут включать сертификацию и контроль процесса производства материала, процесса или агента соответственно.
Вирусная безопасность
В соответствующих случаях в настоящем разделе необходимо представить данные по оценке риска с позиций потенциальной вирусной контаминации. Документация должна соответствовать требованиям, изложенным в главе 3 настоящих Правил.
Прочие посторонние агенты
Подробную информацию по другим посторонним агентам, например, бактериям, микоплазме и грибам, необходимо представить в соответствующих основных разделах досье.
A.3. Новые вспомогательные вещества
В отношении новых вспомогательных веществ необходимо представить сведения, указанные в разделе S регистрационного досье в формате общего технического документа (Модуль 3, 3.2.S), в соответствии с клинической фазой.
A.4. Растворители для восстановления и разбавители
Информация по растворителям для разведения и разбавителям должна быть представлена согласно разделу P регистрационного досье.
4. Существенные поправки
Ниже приводится неисчерпывающий перечень поправок, рассматриваемых в качестве "существенных":
изменение производителя (производителей) активной фармацевтической субстанции или лекарственного препарата;
существенные изменения процесса производства (например, новая экспрессирующая клеточная линия, добавление или исключение стадии очистки, изменение стадий, влияющих на очистку от вирусов, любые процедуры переработки, не указанные в досье исследуемого лекарственного препарата);
изменения, приводящие к образованию новых примесей и родственных соединений;
изменение спецификации, характеризующиеся расширением критериев приемлемости, или исключением, или заменой аналитических методик;
изменения в составе, включая изменение концентрации активной фармацевтической субстанции и содержания вспомогательного вещества;
изменение материала первичной упаковки (изменение разновидности материала);
увеличение срока годности, превышающее утвержденную программу стабильности;
увеличение срока годности, не соответствующее согласованной программе изучения стабильности или без предварительно взятого обязательства (в соответствии с разделами S.7 и P.8)
изменения утвержденных рекомендаций по сроку годности готового к применению лекарственного препарата (после вскрытия упаковки).
Тем не менее увеличение срока годности на основании утвержденной программы стабильности обычно не считается существенной поправкой, если:
каждое дополнительное увеличение срока годности превышает утвержденный срок годности не более чем в 2 раза и не более чем на 12 месяцев;
увеличение охвачено и соответствует утвержденной программе изучения стабильности;
в продолжающихся исследованиях стабильности при заданной температуре хранения значимые тенденции или несоответствие спецификации не выявлены;
заявитель обязуется уведомлять уполномоченные органы о непредвиденных проблемах со стабильностью в продолжающемся исследовании (включая значимые тенденции или несоответствие спецификации) и предложить соответствующие корректирующие действия.
Глава 15. Подобные биологические лекарственные препараты
1. Введение
1.1. Регуляторная основа
В случае разработки нового биологического лекарственного препарата и заявления его на регистрацию в качестве "подобного" оригинальному (референтному) лекарственному препарату, применяются настоящие Правила и правила регистрации и экспертизы лекарственных средств для медицинского применения, утверждаемые Комиссией. В целях получения данных, обосновывающих подобие по качеству, безопасности и эффективности нового биологического препарата и выбранного оригинального (референтного) лекарственного препарата, необходимо провести исследования сопоставимости.
1.2. Область применения
Целью настоящей главы является описание концепции "аналогичных" ("подобных") биологических лекарственных препаратов (далее - "биоаналогичные (биоподобные) препараты") и описание основных принципов, которые необходимо применять в целях разработки, исследований и регистрации.
В целях получения более подробных сведений разработчикам биоаналогичных (биоподобных) лекарственных препаратов, рекомендуется обращаться в уполномоченные органы государств-членов для получения консультации по вопросам разработки.
2. Общие положения
Настоящую главу следует рассматривать в совокупности с другими главами настоящих Правил.
3. Основные принципы
3.1. Применение подхода (концепции) биоаналогичности
(биоподобия) или исследование сопоставимости в рамках
оценки биоаналогичности (биоподобия)
(biosimilarity exercise)
Биоаналогичный (биоподобный) лекарственный препарат - биологический лекарственный препарат, содержащий версию активной фармацевтической субстанции зарегистрированного оригинального (референтного) лекарственного препарата. С помощью всесторонних исследований сопоставимости необходимо подтвердить отсутствие клинически значимых отличий от оригинального (референтного) лекарственного препарата по показателям качества, биологической активности, безопасности и эффективности.
Подход (концепция) биоаналогичности (биоподобия) применим к любому биологическому лекарственному препарату. Однако на практике успех разработки биоаналогичного (биоподобного) лекарственного препарата будет зависеть от возможности произвести лекарственный препарат подобный оригинальному (референтному) лекарственному препарату и достоверно подтвердить подобие (сходство) свойств рассматриваемых лекарственных препаратов. Это предполагает всестороннее установление и сравнение физико-химических и биологических характеристик и требует знания по интерпретации любых различий между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами.
Стандартный подход, применяемый к воспроизведенным лекарственным препаратам (подтверждение биоэквивалентности оригинальному (референтному) лекарственному препарату с помощью исследований биодоступности), применимый к большинству лекарственных препаратов, полученных путем химического синтеза, не достаточен для подтверждения подобия (сходства) биологических, в том числе биотехнологических, препаратов вследствие их сложности. В связи с этим необходимо следовать подходу биоаналогичности (биоподобия), основанному на всесторонних исследованиях сопоставимости.
Научные принципы исследований сопоставимости в рамках оценки биоаналогичности (биоподобия) основываются на принципах, применяемых к оценке влияния изменений производственного процесса биологического лекарственного препарата (в соответствии с главой 9.1) на его качество, безопасность, эффективность.
Применимость подхода биоаналогичности к конкретному биологическому лекарственному препарату зависит от доступности современных аналитических методов, использованных производственных процессов, а также наличия клинических моделей для оценки сопоставимости.
Подход биоаналогичности в первую очередь применим в отношении высоко очищенных препаратов, свойства которых могут быть полнее охарактеризованы (например, многие биотехнологические лекарственные препараты). Подход биоаналогичности сложнее применить к другим видам биологических препаратов, свойства которых в силу их происхождения сложнее охарактеризовать, к примеру, биологические субстанции, получаемые с помощью экстракции из биологических источников, и (или) в отношении которых накоплен ограниченный клинический и регуляторный опыт.
Активная фармацевтическая субстанция биоаналогичного (биоподобного) лекарственного препарата должна быть подобна в молекулярном и биологическом отношении активной фармацевтической субстанции оригинального (референтного) лекарственного препарата. Например, одним из видов молекулярного подобия является требование к совпадению аминокислотных последовательностей белков фармацевтических субстанций. При производстве биоаналогичного (биоподобного) лекарственного препарата как правило используется тот же тип клеточной линии. Если при производстве биоаналогичного (биоподобного) препарата используется иной тип клеточной линии, это должно быть обосновано.
Режим дозирования и путь введения биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов должны быть одинаковыми.
Несовпадение с дозировкой, лекарственной формой, составом, вспомогательными веществами или формой выпуска оригинального (референтного) лекарственного препарата требует обоснования. Такое обоснование требует представления дополнительных данных. Никакие различия не должны снижать эффективность и безопасность.
Целенаправленные изменения для повышения эффективности препарата несовместимы с подходом биоаналогичности (например, оптимизация профиля гликозилирования препарата). Необходимо изучить различия, которые могут способствовать повышению безопасности (например, меньшее содержание примесей или более низкая иммуногенность), такие различия могут не противоречить биоаналогичности.
Биоаналогичный (биоподобный) лекарственный препарат в отношении данных по качеству должен соответствовать всем требованиям, содержащимся в модуле 3 части I приложения N 1 к правилам регистрации и экспертизы лекарственных средств для медицинского применения, утверждаемым Комиссией, и удовлетворять техническим требованиям Фармакопеи Союза и всем дополнительным требованиям, например, предусмотренным настоящими Правилами.
Необходимо подтвердить сопоставимую безопасность и эффективность биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов или обосновать ее иным образом в соответствии с требованиями к данным, предусмотренным правилами регистрации и экспертизы лекарственных средств для медицинского применения, утверждаемым Комиссией. Общие технические и класс-специфичные для биоаналогичных (биоподобных) лекарственных препаратов положения рассматриваются в отдельных главах настоящих Правил. При отсутствии класс-специфичных рекомендаций заявителям следует обращаться в уполномоченные органы за научной консультацией.
Если биоаналогичность (биоподобие) была подтверждена в отношении одного показания к применению, то при надлежащем научном обосновании возможна экстраполяция на другие показания к применению оригинального (референтного) лекарственного препарата.
После регистрации регуляторное требование о необходимости повторного подтверждения биоаналогичности оригинальному (референтному) лекарственному препарату отсутствует, например, в связи с изменением процесса производства.
В целях осуществления фармаконадзора и в соответствии с правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией, следует принять все необходимые меры по однозначной идентификации любого биологического лекарственного препарата, являющегося предметом отчета о подозреваемой нежелательной реакции, с указанием его полного торгового наименования и номера серии.
3.2. Выбор оригинального (референтного)
лекарственного препарата
Оригинальный (референтный) лекарственный препарат должен быть зарегистрирован на территории Союза на основании полного регистрационного досье.
В целях получения согласованных данных и выводов на протяжении всей программы исследований сопоставимости по качеству, безопасности и эффективности в ходе разработки биоаналогичного (биоподобного) лекарственного препарата в качестве препарата сравнения следует использовать 1 и тот же оригинальный (референтный) лекарственный препарат, регистрационное досье которого находится у уполномоченного органа государств-членов. В случае если заявителем подано заявление на регистрацию биоподобного лекарственного препарата в референтное государство, в котором не зарегистрирован оригинальный биологический препарат, уполномоченный орган референтного государства имеет право запросить в рамках межгосударственного взаимодействия регистрационное досье оригинального лекарственного препарата у уполномоченного органа государства-члена, в котором зарегистрирован оригинальный биологический препарат с целью проведения оценки в процессе экспертизы лекарственного препарата. Уполномоченные органы и экспертные организации референтного государства и государства-члена, представившего регистрационное досье оригинального лекарственного препарата, обеспечивают конфиденциальность информации, содержащейся в регистрационном досье оригинального лекарственного препарата, в процессе регистрации и экспертизы лекарственных препаратов.
С целью облегчения глобальной разработки биоаналогичных (биоподобных) лекарственных препаратов и во избежание повторения клинических исследований производитель вправе сравнить разрабатываемый им биоаналогичный (биоподобный) лекарственный препарат в определенных клинических исследованиях и in vivo доклинических исследованиях (при необходимости) с серией оригинального лекарственного препарата, зарегистрированного вне территории Союза (например, государства региона Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для медицинского применения). Кроме того, ответственностью производителя является установление соответствия оригинального (референтного) лекарственного препарата, зарегистрированного вне территории Союза и использованного в виде препарата сравнения, оригинальному (референтному) лекарственному препарату, зарегистрированному на территории Союза.
В целях подтверждения сопоставимости в рамках оценки биоаналогичности (биоподобия) по качеству необходимо провести параллельный анализ биоаналогичного (биоподобного) лекарственного препарата (в рамках промышленных серий, полученных с конкретной производственной площадки, включенной в регистрационное досье) и зарегистрированного на территории Союза оригинального (референтного) лекарственного препарата. Однако для создания целевого профиля качества биоаналогичного (биоподобного) лекарственного препарата допускается одновременное использование в качестве препарата сравнения оригинального (референтного) лекарственного препарата, зарегистрированного как на территории Союза, так и вне территории Союза.
Кроме того, если часть клинических и in vivo доклинических исследований, вошедших в программу разработки, проведены с незарегистрированным на территории Союза препаратом сравнения, заявитель должен представить достаточные данные или сведения, чтобы научно обосновать применимость этих сравнительных данных и связать их с оригинальным (референтным) лекарственным препаратом, зарегистрированным на территории Союза. С научной точки зрения к необходимым связующим данным в обязательном порядке относятся результаты аналитических исследований (например, структурные и функциональные данные), в которых сравнивались все три лекарственных препарата (биоаналогичный (биоподобный) лекарственный препарат-кандидат, зарегистрированный в Союзе оригинальный (референтный) лекарственный препарат и незарегистрированный на территории Союза препарат сравнения), они также могут включать результаты связующих клинических ФК- и (или) ФД-исследований всех 3 лекарственных препаратов. Общая приемлемость подобного подхода и вид требуемых связующих данных определяется в индивидуальном для каждого лекарственного препарата порядке и требует заблаговременного обсуждения с уполномоченными органами. Однако конечное признание приемлемости научного обоснования и связующих данных будет произведено лишь в ходе экспертизы регистрационного досье.
В случае отсутствия регистрации оригинального лекарственного препарата на территории Союза уполномоченный орган государства - члена Союза обращается в Экспертный комитет по лекарственным средствам при Комиссии для получения рекомендации по выбору лекарственного препарата сравнения.
3.3. Принципы установления биоаналогичности (биоподобия)
в рамках оценки сопоставимости
Основной принцип программы разработки биоаналогичного (биоподобного) лекарственного препарата заключается в установлении аналогичности между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами наилучшим способом, обеспечивая применимость ранее установленной безопасности и эффективности оригинального (референтного) лекарственного препарата также и к биоаналогичному (биоподобному) лекарственному препарату.
Биоаналогичный (биоподобный) лекарственный препарат должен быть высоко аналогичен оригинальному (референтному) лекарственному препарату по физико-химическим и биологическим характеристикам. Любые выявленные различия необходимо должным образом обосновать с позиций их потенциального влияния на безопасность и эффективность.
На протяжении всей программы разработки рекомендуется применять поэтапный подход, начинающийся со всестороннего установления физико-химических и биологических характеристик. Объем и характер доклинических исследований in vivo и клинических исследований устанавливают в зависимости от качества доказательств, полученных на предыдущем этапе (этапах), включая надежность данных физико-химических, биологических и in vivo доклинических исследований.
Цель клинических исследований заключается в анализе небольших различий, выявленных на предыдущих этапах, и подтверждении сопоставимости клинических свойств биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов. Клинические данные не допускается использовать для обоснования существенных различий между показателями качества.
Если исследования сопоставимости в рамках оценки биоаналогичности (биоподобия) указывают на значимые различия между биоаналогичным (биоподобным) лекарственным препаратом-кандидатом и оригинальным (референтным) лекарственным препаратом, ставя под сомнение подтверждение биоаналогичности, следует начать самостоятельную разработку для составления полного регистрационного досье.
Конечной целью исследований сопоставимости в рамках оценки биоаналогичности (биоподобия) является исключение каких-либо клинически значимых различий между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственным препаратом. Вследствие чего в целях выявления таких различий дизайн, проведение, конечные точки исследования и (или) выбранная целевая популяция должны обеспечить достаточную чувствительность.
В определенных случаях подтверждающие клинические исследования могут не требоваться. Для этого аналогичная (сопоставимая) эффективность и безопасность должны быть однозначно выведены из аналогичности (сопоставимости) физико-химических характеристик, биологической активности (активности) и ФК- и (или) ФД-профилей биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов. Кроме того, необходимо, чтобы профиль примесей и свойства вспомогательных веществ биоаналогичного (биоподобного) лекарственного препарата не вызывали опасений.
Подобные упрощенные подходы следует согласовывать с уполномоченными органами государств-членов.
Глава 15.1. Биоаналогичные (биоподобные) лекарственные
препараты, содержащие в качестве активной фармацевтической
субстанции биотехнологические белки. Вопросы качества
В настоящей главе по биоаналогичным (биоподобным) лекарственным препаратам, содержащим в качестве активной фармацевтической субстанции белки, полученные с использованием биотехнологии, отражены вопросы качества, установление требований к качеству биологического лекарственного препарата, заявляемого как подобный другому, ранее зарегистрированному на территории Союза лекарственному препарату.
В настоящей главе указаны требования относительно производственного процесса, выполнения сравнительных исследований по доказательству подобия качества, с учетом выбора оригинального (референтного) лекарственного препарата (препарата сравнения), аналитических методов, физико-химических характеристик, биологической активности, чистоты и соответствующих параметров показателей качества для спецификации биоаналогичного (биоподобного) лекарственного препарата.
1. Введение
Как указывается в главе 15 настоящих Правил, разработчик вправе начать разрабатывать новый биологический лекарственный препарат, заявляя его в качестве биоаналогичного (биоподобного) лекарственного препарата по качеству, безопасности и эффективности оригинальному (референтному) лекарственному препарату, зарегистрированному на территории Союза. Разработка подобного биологического лекарственного препарата (биоаналога) частично основана на научных данных, полученных в отношении оригинального (референтного) лекарственного препарата, при условии, что аналогичность активной фармацевтической субстанции биоаналога (биоаналогичного (биоподобного) препарата) по физико-химическим и биологическим свойствам по отношению к активной фармацевтической субстанции оригинального (референтного) лекарственного препарата подтверждена.
Производство и контроль качества биоаналогичных (биоподобных) лекарственных препаратов осуществляются по собственному плану разработки путем применения передовых подходов и учета современных данных. Разработку лекарственного препарата необходимо осуществлять в соответствии с актами, входящими в право Союза.
Сравнение биоаналогичного (биоподобного) лекарственного препарата с общедоступным стандартом, например, фармакопейной статьей, для целей сопоставимости является недостаточным. Необходимо подтвердить сходство (подобие) биоаналогичного (биоподобного) лекарственного препарата оригинальному (референтному) лекарственному препарату, зарегистрированному на территории Союза, избранному компанией для разработки биоаналогичного (биоподобного) лекарственного препарата. Поэтому в целях подтверждения того, что профиль биоаналогичного (биоподобного) лекарственного препарата по качеству, эффективности и безопасности соответствует оригинальному (референтному) лекарственному препарату, необходимо провести значительный объем сравнительных исследований.
Предполагается, что производитель, разрабатывающий биоаналогичный (биоподобный) лекарственный препарат, не имеет доступа ко всей информации, которая позволила бы провести исчерпывающее сравнение с оригинальным (референтным) лекарственным препаратом, особенно о процессе производства. Тем не менее подаваемые аналитические данные должны позволить прийти к твердому заключению о физико-химической и биологической аналогичности между оригинальным (референтным) лекарственным препаратом и биоаналогичным (биоподобным) лекарственным препаратом.
Исследования сопоставимости (при правильном проведении) в рамках оценки биоаналогичности (биоподобия) на этапе качества, включая анализ значимых показателей качества с помощью достаточно чувствительных аналитических методов, могут позволить подать заявление о регистрации биоаналогичного (биоподобного) лекарственного препарата. В этом случае, чтобы завершить разработку биоаналогичного (биоподобного) лекарственного препарата, заявителю потребуется реализовать соответствующую программу доклинической и клинической сопоставимости, предусмотренную настоящими Правилами.
2. Область применения
В настоящей главе рассматриваются вопросы качества при подтверждении сопоставимости биоаналогичных (биоподобных) лекарственных препаратов, содержащих белки, полученные с помощью технологии рекомбинантной ДНК, и их производные, с целью обоснования регистрации. Тем не менее, поскольку подход биоаналогичности (биоподобия) применим к любым биологическим лекарственным препаратам, принципы, раскрываемые в настоящей главе, могут быть применимы к другим биологическим препаратам в индивидуальном порядке.
Исследования сопоставимости при изменениях процесса производства определенного лекарственного препарата (то есть изменения в ходе разработке и после регистрации), описанные в главе 9.1 настоящих Правил, в настоящей главе не рассматриваются.
3. Правовая основа
Необходимо представить полное досье по качеству (модуль 3 регистрационного досье), в соответствии с правилами регистрации и экспертизы лекарственных средств для медицинского применения, утверждаемыми Комиссией, которое следует дополнить подтверждением сопоставимости в рамках оценки биоаналогичности (биоподобия) в соответствии с настоящей главой. Заявителям следует учитывать, что исследования сопоставимости биоаналогичного (биоподобного) лекарственного препарата с оригинальным (референтным) лекарственным препаратом являются дополнительным элементом к стандартным требованиям, предъявляемым к досье по качеству. Их результаты требуют отдельного описания в разделе 3.2.P регистрационного досье.
4. Процесс производства подобного биологического
лекарственного препарата
Разработка биоаналогичного (биоподобного) лекарственного препарата и ее документирование должны охватывать 2 различающихся аспекта:
i) молекулярные характеристики и показатели качества целевого профиля препарата должны быть сопоставимы с оригинальным (референтным) лекарственным препаратом;
ii) осуществление и постоянство процесса производства биоаналогичного (биоподобного) лекарственного препарата.
Целевой профиль качества биоаналогичного (биоподобного) лекарственного препарата (далее - ЦПКП) должен быть основан на данных выбранного оригинального (референтного) лекарственного препарата, включая общедоступные сведения и данные, полученные при детальном установлении характеристик оригинального (референтного) лекарственного препарата. ЦПКП должен составлять основу разработки биоаналогичного (биоподобного) лекарственного препарата и процесса его производства. Такой ЦПКП следует рассматривать в качестве инструмента разработки, отдельные целевые диапазоны которого по мере получения новых сведений об оригинальном (референтном) лекарственном препарате могут изменяться в ходе разработки.
Производство биоаналогичного (биоподобного) лекарственного препарата и контроль его качества осуществляются по собственному плану разработки с учетом современных сведений о производственных процессах и последствий для свойств препарата. Как в случае любого биологического лекарственного препарата, биоаналогичный (биоподобный) лекарственный препарат характеризуется молекулярной композицией активной фармацевтической субстанции, образующейся в результате процесса ее производства, привносящего свои молекулярные варианты, изоформы и другие родственные вещества, а также производственные примеси. Как следствие, в целях достижения ЦПКП процесс производства необходимо должным образом спроектировать. Необходимо тщательно выбрать экспрессирующую систему, учитывая различия между экспрессирующими системами, которые могут привести к нежелательным последствиям, таким как атипичный профиль гликозилирования, повышенная вариабельность в содержании примесей и (или) иной по сравнению с оригинальным (референтным) лекарственным препаратом профиль примесей.
При выборе состава биоаналогичного (биоподобного) лекарственного препарата необходимо принимать во внимание современные технологии, идентичность составу оригинального (референтного) лекарственного препарата не является обязательной. Независимо от выбранного состава, необходимо подтвердить обоснованность предлагаемого состава с точки зрения его стабильности, сопоставимости (взаимодействия со вспомогательными веществами, растворителями и первичными упаковочными материалами), сохранности, активности и количественного содержания активной фармацевтической субстанции. Если состав и (или) система "контейнер - укупорка" отличаются от таковых оригинального (референтного) лекарственного препарата (включая любой материал, контактирующий с лекарственным препаратом), их потенциальное влияние на эффективность и безопасность биоаналогичного (биоподобного) лекарственного препарата необходимо должным образом обосновать.
Стабильность биоаналогичного (биоподобного) лекарственного препарата определяется в соответствии с главой 8 настоящих Правил. Любые утверждения относительно стабильности и совместимости необходимо обосновать собственными экспериментальными данными, они не могут быть экстраполированы на основании данных об оригинальном (референтном) лекарственном препарате.
Поскольку биоаналогичный (биоподобный) лекарственный препарат имеет собственный жизненный цикл, при внесении изменений в производство (активной фармацевтической субстанции и (или) лекарственного препарата) в ходе разработки, необходимо провести оценку сопоставимости в соответствии с главой 9.1 настоящих Правил. В целях упрощения экспертизы регистрационного досье все исследования сопоставимости изменений процесса, произведенных в ходе разработки, необходимо четко обозначить и представить отдельно от исследований сопоставимости для подтверждения биоаналогичности с оригинальным (референтным) лекарственным препаратом. В ходе разработки биоаналогичного (биоподобного) лекарственного препарата могут быть внесены изменения в процесс производства, однако при получении необходимых данных по качеству, безопасности и эффективности для подтверждения биоаналогичности по отношению к оригинальному (референтному) лекарственному препарату, строго рекомендуется использовать лекарственный препарат, произведенный в рамках промышленного процесса производства, отражающего профиль качества серий, выпускаемых в реализацию.
5. Исследование сопоставимости с оригинальным (референтным)
лекарственным препаратом. Вопросы качества
5.1. Оригинальный (референтный) лекарственный препарат
Необходимо четко обозначить оригинальный (референтный) лекарственный препарат, использованный в исследованиях сопоставимости в рамках оценки биоаналогичности (биоподобия) на этапе качества (например, торговое наименование, лекарственная форма, состав, дозировка, происхождение лекарственного препарата, номера серий, номер партии, дата производства серий, назначение). В целях формирования репрезентативного профиля качества и получения достоверных данных по сопоставимости необходимо использовать несколько серий оригинального (референтного) лекарственного препарата. При наличии нескольких дозировок или лекарственных форм их выбор необходимо должным образом обосновать. При определении целевого профиля качества необходимо учитывать "возраст" (по отношению к сроку годности) различных серий оригинального (референтного) лекарственного препарата.
Для подтверждения биоаналогичности в качестве оригинального (референтного) лекарственного препарата не могут быть использованы общедоступные стандартные образцы (например, фармакопейные). Однако как указано в подразделе 5.3 настоящей главы, применение общедоступных стандартных образцов играет важную роль в квалификации и стандартизации методов.
5.2. Исследование сопоставимости в рамках оценки
биоаналогичности (биоподобия)
В целях подтверждения того, что профиль качества биоаналогичного (биоподобного) лекарственного препарата высоко аналогичен оригинальному (референтному) лекарственному препарату требуется провести обширные исследования сопоставимости. Они должны включать всесторонний анализ предложенного биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата с использованием чувствительных ортогональных методов для выявления не только сходств, но и потенциальных различий между показателями качества. Этот анализ, если не обосновано обратное, должен включать параллельные сравнительные исследования. Любые обнаруженные различия между показателями качества необходимо должным образом обосновать с позиций их потенциального влияния на безопасность и эффективность.
Если подтвердится наличие значимых различий по качеству (в отношении которых невозможно обосновать отсутствие их клинически значимого влияния), заявление разработчика об аналогичности по отношению к оригинальному (референтному) лекарственному препарату вызовут сомнения. В таких случаях целесообразно рассмотрение вопроса о регистрации препарата на основе полного регистрационного досье. В качестве альтернативы заявитель вправе принять решение о пересмотре процесса производства в целях минимизации или предотвращения этих различий.
Цель исследований сопоставимости в рамках оценки биоаналогичности (биоподобия) заключается в подтверждении высокой степени аналогичности (подобия) биоаналогичного (биоподобного) лекарственного препарата и выбранного заявителем оригинального (референтного) лекарственного препарата на уровне готовой лекарственной формы. Не требуется, чтобы все показатели качества биоаналогичного (биоподобного) лекарственного препарата полностью совпадали с показателями качества оригинального (референтного) лекарственного препарата. Однако если обнаружены качественные и (или) количественные различия, их необходимо обосновать и при необходимости подтвердить отсутствие их влияния на клинические характеристики препарата. Как указано в главах 15 и 15.2 настоящих Правил, это может потребовать представления дополнительных доклинических и (или) клинических данных. Особое внимание необходимо уделить показателям качества, которые могут повлиять на иммуногенность или активность, или не выявленным у оригинального (референтного) лекарственного препарата.
Заявитель должен подтвердить, что целевой продукт (включая родственные соединения), содержащийся в биоподобном лекарственном препарате, аналогичен продукту оригинального (референтного) лекарственного препарата. В противоположность этому, производственные примеси оригинального (референтного) и биоаналогичного (биоподобного) лекарственных препаратов могут различаться, однако эти различия необходимо минимизировать. Предпочтительно полагаться на процессы очистки, направленные на удаление примесей, чем реализацию доклинической программы испытаний в целях их квалификации. Различия, которые могут обладать преимуществами с точки зрения безопасности (например, меньшее содержание примесей), необходимо объяснить, однако они не станут препятствием для установления биоаналогичности (биоподобия).
По возможности для исследований сопоставимости в рамках оценки биоаналогичности (биоподобия) следует установить численные диапазоны показателей качества. Эти диапазоны должны преимущественно основываться на экспериментально определенных диапазонах показателей качества оригинального (референтного) лекарственного препарата и если не обосновано обратное не должны быть шире, чем вариабельность его репрезентативных серий. Выбранные диапазоны необходимо обосновать с учетом числа испытанных партий оригинального (референтного) лекарственного препарата, изученных показателей качества, "возраста" серий на момент испытаний и использованной аналитической методики. При обосновании в целях определения диапазонов показателей качества допускается использовать методы описательной статистики. Допустимые диапазоны, используемые в исследованиях сопоставимости в рамках оценки биоаналогичности (биоподобия) с оригинальным (референтным) лекарственным препаратом, следует рассматривать отдельно от диапазонов, указанных в спецификациях на выпуск в соответствии с разделом 6 настоящей главы.
Поскольку процесс производства оригинального (референтного) лекарственного препарата протекает по собственному жизненному циклу, возможно возникновение различий по некоторым показателям качества. Такие случаи могут произойти в период разработки биоаналогичного (биоподобного) лекарственного препарата и ЦПКП не будет отражать оригинальный (референтный) лекарственный препарат, находящийся на рынке. Диапазоны, установленные до и после обнаруженного сдвига профиля качества, как правило, допускается использовать для обоснования исследований сопоставимости в рамках оценки биоаналогичности (биоподобия) на этапе качества, поскольку любой из диапазонов отражает свойства оригинального (референтного) лекарственного препарата. Значения показателей качества, находящихся вне или между диапазоном, установленным для показателя качества оригинального (референтного) лекарственного препарата, необходимо должным образом обосновать с позиций их потенциального влияния на безопасность и эффективность. Следует также отметить, что регуляторное требование о повторном подтверждении биоаналогичности после регистрации отсутствует.
5.3. Аналитические вопросы
В целях достоверного подтверждения сопоставимости биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата по качеству необходимо провести параллельные исследования по установлению характеристик биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата с учетом современных достижений.
Обязанностью заявителя является подтверждение способности методов, выбранных в целях исследований сопоставимости в рамках оценки биоаналогичности (биоподобия), обнаружить незначительные различия между всеми параметрами, влияющими на оценку качества (например, способность с высокой чувствительностью обнаруживать значимые варианты). Методы (методики), использованные в исследованиях по установлению характеристик, являются неотъемлемой частью комплекта данных по качеству и требуют надлежащей квалификации (подтверждения применимости) для целей изучения сопоставимости. Если применимо, в целях квалификации и стандартизации методов необходимо использовать стандартные образцы и материалы (например, фармакопейные, ВОЗ).
Прямой или параллельный анализ биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата с помощью некоторых аналитических методик может оказаться невозможным или быть малоинформативным (например, вследствие низкой концентрации активной фармацевтической субстанции и (или) наличия мешающих вспомогательных веществ (таких как альбумин), которые искажают или не позволяют получить адекватные результаты). В этом случае исследуемый образец может быть приготовлен из готовой лекарственной формы с использованием подходящих методов (например, экстракции, концентрирования и (или) других). В таких ситуациях необходимо описать методики пробоподготовки, их влияние на пробы необходимо должным образом документировать и проанализировать (например, путем сравнения активных фармацевтических субстанций до и после получения готовой лекарственной формы с последующим извлечением из готовой лекарственной формы).
5.3.1. Физико-химические свойства.
Сравнение физико-химических свойств включает не только оценку соответствующих параметров, но и установление структуры родственных соединений и родственных примесей. В программе установления физико-химических характеристик необходимо предусмотреть определение состава, физических свойств, первичной структуры и структур более высокого порядка биоаналогичного (биоподобного) лекарственного препарата с применением соответствующих методов. Необходимо подтвердить целевую аминокислотную последовательность биоаналогичного (биоподобного) лекарственного препарата, которая не должна отличаться от последовательности оригинального (референтного) лекарственного препарата. В соответствующих случаях необходимо сопоставить N- и C-концевые аминокислотные последовательности, свободные SH-группы и дисульфидные мостики. Все модификации и (или) укорочения необходимо оценить количественно и описать внутренне присущую (собственную) или обусловленную экспрессирующей системой вариабельность. Любые выявленные различия между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами необходимо обосновать с позиций профиля микрогетерогенности оригинального (референтного) лекарственного препарата (например, вариабельностью C-концевого лизина).
Необходимо должным образом охарактеризовать наличие и степень посттрансляционных модификаций (например, гликозилирования, окисления, дезамидирования, укорочения). Необходимо тщательно сравнить углеводные структуры (при их наличии), включая общий гликановый профиль, сайт-специфичные профили гликозилирования, в том числе занятость сайтов гликозилирования. Наличие структур или вариантов гликозилирования, отсутствующих в оригинальном (референтном) лекарственном препарате, может вызвать возражения и требует надлежащего обоснования, уделяя особое внимание структурам, нехарактерным для человека (нехарактерные для человека ковалентные связи, последовательности или сахара).
5.3.2. Биологическая активность.
В исследованиях сопоставимости в рамках оценки биоаналогичности (биоподобия) необходимо предусмотреть сравнительную оценку биологических свойств биоаналога (биоподобного препарата) и оригинального (референтного) лекарственного препарата как неотъемлемого этапа установления всестороннего профиля характеристик. Биологическая активность - это особая способность или свойство препарата оказывать определенный биологический эффект. В целях определения биологической активности необходимо использовать соответствующие биологические методы количественного определения, основанные на различных взаимодополняющих принципах. В зависимости от биологических свойств препарата допускается использовать различные способы количественного определения (например, количественное определение связывания с лигандом или рецептором, ферментные методы, методы на основе клеток и функциональные методы), принимая во внимание их ограничения. В целях преодоления ограничений, обусловленных валидационными характеристиками отдельных количественных биологических методов, рекомендуется придерживаться ортогональных (взаимодополняющих) подходов. Если применимо, необходимо использовать отдельные методы для оценки связи и активации рецепторов. В соответствующих случаях допускается приводить ссылки на доклинический и (или) клинический разделы досье. Необходимо подтвердить, что количественные биологические методы чувствительны, специфичны и обладают достаточной дискриминационной (отличительной) способностью. По возможности, результаты соответствующего биологического метода следует представлять в калиброванных (градуированных) по международным или национальным стандартным образцам (при наличии) единицах активности. Если применимо, эти методы должны удовлетворять соответствующим требованиям Фармакопеи Союза к биологическим методам количественного определения.
5.3.3. Иммунохимические свойства.
Как указано в главе 15.3 настоящих Правил, иммунологические функции моноклональных антител и производных от них веществ (например, гибридные белки на основе Fc-фрагментов IgG) необходимо всесторонне сравнить. Это обычно включает сравнение аффинности продуктов (целевого продукта, родственных соединений и родственных примесей) к их мишеням. Кроме того, если не обосновано обратное, необходимо сравнить аффинность Fc-фрагментов к соответствующим рецепторам (например, FcyR, C1q, FcRn). В целях сравнения способности индуцировать Fab- и Fc-ассоциированные эффекторные функции необходимо использовать соответствующие методы.
5.3.4. Чистота и примеси.
Используя комбинацию аналитических методик, необходимо провести качественное и количественное сравнения профилей чистоты и примесей биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов. В целях идентификации и сравнения родственных соединений и примесей необходимо использовать подходящие ортогональные и современные методы. При таком сравнении необходимо принимать во внимание характерные пути деградации (например, окисление, дезамидирование, агрегация) биоаналогичного (биоподобного) лекарственного препарата и потенциальные посттрансляционные модификации белков. Необходимо указать "возраст" и срок годности оригинального (референтного) лекарственного препарата на момент проведения испытаний. В соответствующих случаях необходимо проанализировать его потенциальное влияние на профиль качества. В целях достоверного подтверждения аналогичности путей деградации оригинального (референтного) лекарственного препарата и биоаналогичного (биоподобного) лекарственного препарата следует сравнить значимые показатели качества, испытанные в определенные временные точки и при определенных условиях хранения (например, ускоренных или стрессовых).
Производственные примеси (например, белки клетки-хозяина, ДНК клетки-хозяина, реагенты, примеси, обусловленные последующей после культивирования обработкой и т.д.) будут отличаться от процесса к процессу. В связи с этим качественное сравнение указанных параметров в исследованиях сопоставимости в рамках оценки биоаналогичности (биоподобия) может быть не значимо. Тем не менее, следуя действующим рекомендациям и фармакопейным требованиям, следует использовать современные аналитические методы, а потенциальные риски, обусловленные этими обнаруженными примесями (например, иммуногенность), необходимо должным образом документировать и обосновывать.
5.3.5. Количественное содержание.
Количественное содержание следует определять, используя соответствующие методы, и выражать в тех же единицах, что и у оригинального (референтного) лекарственного препарата. Необходимо подтвердить сопоставимость количественного содержания биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов.
6. Спецификации
Подобно всякому биотехнологическому препарату выбор испытаний, включаемых в спецификации (или стратегии контроля) как фармацевтической субстанции, так и лекарственного препарата зависит от его характеристик и должен осуществляться в соответствии с главой 6 настоящих правил. Необходимо представить основания, руководствуясь которыми был выбран предлагаемый диапазон критериев приемлемости для рутинных испытаний.
Заявленный срок годности препарата должен быть обоснован полными данными по стабильности биоаналогичного (биоподобного) лекарственного препарата. Сравнительные исследования стабильности в реальном времени и реальных условиях между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами не требуются.
Глава 15.2. Биоаналогичные (биоподобные) лекарственные
препараты, содержащие в качестве активной фармацевтической
субстанции биотехнологические белки. Вопросы доклинических
и клинических исследований
В настоящей главе рассматриваются общие принципы доклинического и клинического этапов разработки и оценки регистрационных досье подобных биоаналогичных (биоподобных) лекарственных препаратов, содержащих в качестве активной фармацевтической субстанции рекомбинантные белки.
В главе изложены требования к данным, получаемым в ходе доклинических и клинических исследований лекарственного препарата, заявленного в качестве биоаналогичного (биоподобного) лекарственного препарата.
В разделе по доклиническим исследованиям приведена информация о фармако-токсикологической оценке, а в разделе по клиническим исследованиям изложены требования к исследованиям фармакокинетики, фармакодинамики и эффективности. В разделе по клинической безопасности и фармаконадзору рассматриваются исследования клинической безопасности, включая иммуногенность, а также план управления рисками. Рекомендуется придерживаться пошагового подхода к проведению доклинических и клинических исследований.
1. Введение
Биоаналогичный (биоподобный) лекарственный препарат - биологический лекарственный препарат, содержащий версию активной фармацевтической субстанции (действующего вещества) зарегистрированного оригинального (референтного) лекарственного препарата, для которого продемонстрировано сходство (подобие) с оригинальным (референтным) лекарственным препаратом. С помощью всесторонних исследований сопоставимости необходимо подтвердить отсутствие клинически значимых отличий от оригинального (референтного) лекарственного препарата по показателям качества, биологической активности, безопасности и эффективности.
Регистрационное досье биоаналогичного (биоподобного) лекарственного препарата должно содержать полное досье по качеству вместе с данными, подтверждающими сопоставимость с оригинальным (референтным) лекарственным препаратом, полученными при проведении надлежащих физико-химических и in vitro биологических испытаний, доклинических и клинических исследований.
Вопросы качества, значимые для подтверждения сопоставимости в рамках оценки биоаналогичности (биоподобия), рассмотрены в главе 15.1 настоящих Правил.
Природа и сложность строения оригинального (референтного) лекарственного препарата влияют на объем доклинических или клинических исследований для подтверждения биоаналогичности. Различия, обнаруженные по результатам физико-химических и биологических испытаний, задают направление планированию доклинических и клинических исследований. К прочим факторам, требующим учета, относятся механизм действия (например, вовлеченный рецептор) по всем зарегистрированным показаниям к применению оригинального (референтного) лекарственного препарата и патогенетические механизмы заболеваний, утвержденных в качестве показаний к применению (например, общие механизмы между различными показаниями к применению), а также иммуногенность оригинального (референтного) лекарственного препарата.
Заявитель должен проанализировать результаты изучения оригинального (референтного) лекарственного препарата на предмет прогностической ценности in vitro испытаний и животных моделей, а также корреляции между дозой (экспозицией) и фармакодинамикой. Кроме того, заявитель должен изучить корреляцию между фармакодинамикой и клиническим ответом. Наличие подходящих биомаркеров может сократить доклиническую и клиническую разработку. Профиль безопасности оригинального (референтного) лекарственного препарата будет основным объектом внимания исследований клинической безопасности как на предрегистрационном, так и пострегистрационном этапе.
Если исследования сопоставимости в рамках оценки биоаналогичности (биоподобия) указывают на наличие значимых различий между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом сравнения, ставя под сомнение подтверждение биоаналогичности (биоподобия), то такой лекарственный препарат не может быть зарегистрирован в качестве биоаналогичного (биоподобного) и следует начать самостоятельную разработку для составления полного регистрационного досье в соответствии с главой 15 настоящих Правил.
2. Область применения
В настоящей главе рассмотрены общие принципы доклинической и клинической разработки и оценки регистрационных досье биоаналогичных (биоподобных) лекарственных препаратов, содержащих в качестве активной фармацевтической субстанции биотехнологические белки. Тем не менее принципы, изложенные в настоящей главе, могут быть применимы к другим биологическим лекарственным препаратам в индивидуальном порядке. Исследования сопоставимости при изменениях процесса производства определенного препарата (изменения в ходе разработке и после регистрации) в настоящей главе не рассматриваются.
3. Связь с другими главами
В главах 15.3 - 15.11 настоящих Правил содержатся класс-специфичные указания, облегчающие доклиническую и клиническую разработку биоаналогичных (биоподобных) лекарственных препаратов в определенных областях.
4. Доклинические исследования
В целях обоснования биоаналогичности перед началом клинических исследований необходимо провести соответствующие доклинические исследования. При оценке аналогичности биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата рекомендуется придерживаться пошагового подхода. Сначала следует провести аналитические исследования (глава 15.2 настоящих Правил) и in vitro фармако-токсикологические исследования, чтобы затем принять решение о необходимом объеме исследований на животных, при необходимости проведения таких исследований.
Важно отметить, что для составления надлежащей программы доклинических исследований требуется четкое понимание характеристик оригинального (референтного) лекарственного препарата. Результаты физико-химических и биологических испытаний по установлению характеристик (биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов) необходимо проанализировать с точки зрения влияния на эффективность и безопасность.
Предлагается следующий подход, который следует применять к рассматриваемому препарату в индивидуальном порядке. Принятый подход необходимо всесторонне обосновать в доклиническом обзоре регистрационного досье (модуль 2.4 регистрационного досье).
4.1. Шаг 1. Исследования in vitro
В целях оценки любого потенциального различия биологической активности между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами необходимо представить результаты сравнительных исследований in vitro, некоторые из которых могут быть доступны по итогам испытаний качества.
Такие исследования должны включать следующее:
испытание связывания с мишенью (например, рецепторами, антигенами, ферментами), известной своей вовлеченностью в фармако-токсикологические эффекты и (или) фармакокинетику оригинального (референтного) препарата;
испытание передачи сигнала и функциональной активности или жизнеспособности клеток, известных своей значимостью для фармако-токсикологических эффектов оригинального (референтного) лекарственного препарата.
Исследования должны быть сравнительными, а не направленными исключительно на оценку ответа per se. Для получения однозначных результатов методы должны быть научно обоснованными и пригодными для своего назначения.
Исследования должны быть чувствительными, специфичными и обладать достаточной дискриминативной способностью, чтобы подтвердить, что наблюдаемые различия в показателях качества клинически незначимы. В исследованиях необходимо сравнить зависимость "концентрация-активность (связывание)" биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов с фармакологической мишенью, охватив диапазон концентраций, в котором потенциальные различия могут быть выявлены с наибольшей чувствительностью.
Исследования следует провести на достаточном числе серий оригинального (референтного) лекарственного препарата и биоаналогичного (биоподобного) лекарственного препарата, отражающего свойства препарата, предназначенного для клинического применения. На необходимое число серий влияют вариабельность испытания и межсерийная вариабельность. Испытуемого числа серий должно быть достаточно, чтобы составить полноценное заключение о вариабельности конкретного параметра как биоаналогичного (биоподобного) лекарственного препарата, так и оригинального (референтного) лекарственного препарата, и аналогичности обоих лекарственных препаратов.
Все эти испытания должны охватывать весь спектр фармакологических и токсикологических аспектов, известных своей клинической значимостью для оригинального (референтного) лекарственного препарата и соответствующего класса препаратов.
Заявитель должен проанализировать степень репрезентативности (прогностической ценности) использованных in vitro испытаний для клинической ситуации в соответствии с актуальными научными знаниями.
Поскольку in vitro испытания могут быть более специфичны и чувствительны в выявлении различий между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом, чем исследования у животных, их следует рассматривать в качестве первостепенных (основополагающих) в доклинических исследованиях сопоставимости в рамках оценки биоаналогичности (биоподобия).
4.2. Шаг 2. Определение необходимости проведения
in vivo исследований
Белки, полученные с помощью биотехнологических методов, могут опосредовать эффекты in vivo, которые нельзя полностью охарактеризовать в исследованиях in vitro. Следовательно, в целях представления недостающих сведений может потребоваться доклиническая оценка в исследованиях in vivo при условии наличия релевантной in vivo модели (с точки зрения вида животных и дизайна исследования).
Факторы, принимаемые во внимание при оценке необходимости доклинических исследований in vivo, включают перечисленные ниже, но не ограничиваются ими:
наличие потенциально значимых показателей качества, которые не выявлены у оригинального (референтного) лекарственного препарата (например, новые посттрансляционные модификации);
наличие потенциально значимых количественных различий в показателях качества между рассматриваемым биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами;
значимые различия в составе, например, наличие вспомогательных веществ, редко используемых вместе с биотехнологическими белками.
Несмотря на то, что указанные выше факторы, взятые по отдельности, необязательно требуют проведения испытаний in vivo, необходимо проанализировать эти факторы в комплексе, чтобы оценить степень опасений и необходимость проведения in vivo испытаний.
Если исследования сопоставимости в рамках оценки биоаналогичности (биоподобия) физико-химических и биологических характеристик и доклинические исследования in vitro (см. шаг 1) признаны удовлетворительными, а на шаге 2 вопросов, которые могли бы препятствовать непосредственному переходу к клиническим исследованиям, не возникает, исследования на животных in vivo, как правило, не требуются.
Если присущие препарату факторы, влияющие на ФК и (или) биораспределение, например, выраженное гликозилирование, на уровне качества или in vitro охарактеризовать в достаточной степени невозможно, могут потребоваться исследования in vivo. Заявитель должен подробно проанализировать, следует ли проводить подобные исследования на животных или в качестве отдельного этапа клинической разработки, например, у здоровых добровольцев.
При необходимости получения дополнительных данных in vivo необходимо принимать во внимание доступность релевантных видов животных или других релевантных моделей (например, трансгенных животных, трансплантатных моделей).
Если релевантная in vivo животная модель отсутствует, заявитель вправе начать исследования у человека, принимая во внимание принципы снижения любого потенциального риска.
4.3. Шаг 3. Исследования in vivo
Если in vivo оценка сочтена заявителем необходимой, цель исследования (ФК, и (или) ФД, и (или) безопасности) зависит от требуемых дополнительных сведений. Исследования на животных следует спланировать таким образом, чтобы получить максимально полные сведения. При планировании любого in vivo исследования необходимо учитывать принцип 3R (замена, улучшение, сокращение) (replacement, refinement, reduction). В зависимости от используемых конечных точек, в конце исследования может не потребоваться умерщвление животных. Необходимо обосновать продолжительность исследования (включая период наблюдения), принимая во внимание ФК-свойства оригинального (референтного) лекарственного препарата и его клиническое применение.
Если позволяет модель и в отсутствие обоснования иного, количественному сравнению подлежат ФК и ФД биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов, включая, если выполнимо, оценку зависимости "доза-эффект", в том числе предполагаемую экспозицию у человека.
В отношении исследований безопасности следует придерживаться гибкого подхода, в особенности если единственными релевантными видами животных являются нечеловекообразные приматы. Проведение стандартных исследований токсичности при многократном введении у нечеловекообразных приматов, как правило, не рекомендуется. При достаточном обосновании можно провести исследование токсичности при многократном введении с измененным дизайном (например, используя лишь одну дозу биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата и (или) лишь один пол и (или) исключив группу восстановления) или прижизненную оценку параметров безопасности (таких как клинические признаки, масса тела и витальные функции). Если в исследованиях токсичности при многократном введении изучается только одна доза, она должна быть близка к верхней границе диапазона дозирования, ее следует обосновывать с позиций ожидаемой токсичности оригинального (референтного) лекарственного препарата.
Проведение токсикологических исследований на нерелевантных видах животных (например, с целью оценки исключительно неспецифической токсичности, обусловленной примесями) не рекомендуется. Вследствие различий в процессах, используемых производителями биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов, могут возникать качественные различия в производственных примесях (например, белках клетки-хозяина). Содержание таких примесей должно быть минимально, что и является наилучшей стратегией минимизации любого ассоциированного риска.
Качественные или количественные различия в родственных вариантах (например, характере гликозилирования, вариантах, отличающихся зарядами) могут влиять на биологические функции биотехнологического белка, их следует оценить в надлежащих испытаниях in vitro. Эти различия и примеси могут влиять на иммуногенный потенциал и потенциал развития гиперчувствительности. Эти эффекты с помощью исследований на животных тяжело прогнозируются и требуют дальнейшей оценки в клинических исследованиях.
Оценка иммуногенности у животных, в целом, не прогнозирует иммуногенность у человека, но она может потребоваться для интерпретации in vivo исследований на животных (в соответствии с главой 5.4 настоящих Правил). Следовательно, необходимо осуществлять сбор образцов крови и хранить их для будущего анализа фармакокинетических (токсикокинетических) данных, если в дальнейшем он потребуется.
При доклиническом испытании биоаналогичных (биоподобных) лекарственных препаратов исследования фармакологической безопасности, репродуктивной токсичности и канцерогенности не требуются.
Исследования местной переносимости, как правило, не требуются. Однако, если препарат содержит вспомогательные вещества, для которых опыт использования при рассматриваемом пути введения отсутствует или мал, может потребоваться оценка местной переносимости. Если проводятся другие исследования in vivo, оценку местной переносимости можно включить в их дизайн, вместо того чтобы проводить отдельные исследования.
5. Клинические исследования
Технология производства биоаналогичного (биоподобного) препарата в процессе разработки будет подвергаться оптимизации. Однако клинические данные, необходимые для анализа сопоставимости в рамках оценки биоаналогичности (биоподобия), рекомендуется получать, используя биоаналогичный (биоподобный) лекарственный препарат, полученный с помощью коммерческого процесса производства и, следовательно, отражающий профиль качества серий, которые будут введены в реализацию. Любые отклонения от этого требования следует обосновать и подкрепить с помощью необходимых дополнительных связующих данных (в соответствии с главой 9.1 настоящих Правил).
Клинический анализ сопоставимости в рамках оценки биоаналогичности (биоподобия), как правило, пошаговый процесс, который следует начинать с ФК-исследований и, если выполнимо, ФД-исследований, с последующим проведением исследования эффективности и безопасности или, в определенных случаях, подтверждающих ФК-исследований (ФД-исследований) для демонстрации сопоставимости клинической эффективности и безопасности в рамках оценки биоаналогичности (биоподобия).
5.1. Фармакокинетические исследования
Сравнительные ФК-исследования, направленные на подтверждение аналогичности ФК-профиля биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов с позиций ключевых ФК-параметров, являются неотъемлемой частью программы разработки биоаналогичного (биоподобного) лекарственного препарата.
Дизайн ФК-исследования зависит от различных факторов, включая клинический контекст, безопасность, ФК-характеристики оригинального (референтного) препарата (мишень-опосредованные распределение, метаболизм и элиминация, линейность и нелинейность ФК, временную зависимость, период полувыведения и т.п.), описанных в правилах исследования биоэквивалентности лекарственных препаратов в рамках Союза, утверждаемых Комиссией и иных актах, входящих в право Союза. Биоаналитические методики должны соответствовать своему целевому назначению и быть валидированы в соответствии с правилами исследования биоэквивалентности лекарственных препаратов в рамках Союза.
Перед началом исследования необходимо задать и обосновать границы сопоставимости основных фармакокинетических параметров в рамках оценки биоаналогичности (биоподобия). В отсутствие специальных критериев обоснованной отправной точкой для планирования сравнительных фармакокинетических исследований биологических лекарственных препаратов могут служить критерии, используемые в стандартных исследованиях биоэквивалентности, изначально разработанные для лекарственных препаратов для приема внутрь, действующие вещества которых получены химическим путем. Однако в отличие от небольших молекул интерпретация исследований биоэквивалентности биологических лекарственных препаратов менее однозначна. В первом случае молекулы признаются идентичными, тогда как для биологических лекарственных препаратов ФК используется с целью обнаружения различий во взаимодействии оригинального (референтного) и биоаналогичного (биоподобного) лекарственных препаратов с организмом. Это значит, что выдерживание 90 процентных ДИ отношений биоаналогичным (биоподобным) лекарственным препаратом к оригинальному (референтному) лекарственному препарату в пределах заранее установленного обоснованного диапазона приемлемости само по себе может быть недостаточным. При интерпретации аналогичности следует также учитывать расположение и ширину доверительного интервала. Например, потребуется объяснить и обосновать в качестве не препятствующего биоаналогичности диапазон приемлемости статистически значимых различий в 90 процентных ДИ соответствующих фармакокинетических параметров. С другой стороны, если 90 процентный ДИ пересекает заранее установленные границы, заявитель должен объяснить подобные различия и установить их причину. В индивидуальном порядке допускается коррекция на содержание белка, если она заранее предусмотрена и должным образом обоснована с включением результатов испытания оригинального (референтного) и биоаналогичного (биоподобного) лекарственных препаратов в протокол.
Несмотря на то что мишень-опосредованный клиренс представляет большую важность в исследованиях биоаналогичности, его изучение при выраженной вариабельности экспрессии мишени, включая вариабельность во времени, может оказаться невыполнимым. Поскольку ожидается, что исследования in vitro покажут сопоставимое взаимодействие между биоаналогичным (биоподобным) лекарственным препаратом и его мишенью (включая FcRn для МкАТ), отсутствие опорного ФК-исследования у целевой популяции допустимо, если дополнительные ФК-данные собраны в исследованиях эффективности, безопасности и (или) ФД-исследованиях, поскольку это позволит более глубоко изучить клиническое влияние вариабельной фармакокинетики и возможные изменения ФК во времени. Этого можно достичь, определяя ФК-профиль у подгруппы пациентов или с помощью популяционной фармакокинетики.
Предпочтительно проведение перекрестного исследования с однократным введением с полным описанием ФК-профиля, включая фазу поздней элиминации. В отношении веществ с длительным периодом полувыведения и (или) высоким риском иммуногенности могут потребоваться параллельные исследования. Дозы в ФК-исследовании сопоставимости в рамках оценки биоаналогичности (биоподобия) с однократным введением у здоровых добровольцев могут быть ниже, чем рекомендуемые терапевтические дозы. ФК-исследования у здоровых добровольцев не всегда выполнимы. В этом случае если исследование с однократным введением не выполнимо, ФК необходимо изучить у пациентов в качестве этапа исследования с многократным введением. Необходимо выбрать чувствительную модель (популяцию), то есть такую, которая обладает меньшим количеством факторов, вызывающих выраженную межиндивидуальную или зависящую от времени вариабельность.
Если оригинальный (референтный) лекарственный препарат вводится внутривенно и подкожно, изучения подкожного пути введения, как правило, достаточно, поскольку он охватывает как абсорбцию, так и элиминацию. Таким образом, допускается не проводить оценку внутривенного введения, если сопоставимость в рамках оценки биоаналогичности (биоподобия) при подкожном пути введения подтверждена как для абсорбции, так и для элиминации. Отказ от проведения ФК-исследования при внутривенном введении необходимо обосновать, например, тем, что константа абсорбции молекулы гораздо меньше константы элиминации (инвертированная кинетика).
Основными параметрами ФК-исследования с однократным введением являются 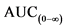 при внутривенном введении и
при внутривенном введении и 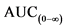 и, как правило, Cmax - при подкожном. Необходимо также оценить такие вторичные параметры, как tmax, объем распределения и период полувыведения. Основными параметрами в исследовании при многократном введении являются AUC, усеченная с момента первого введения до второго введения (AUC0-t), и AUC на протяжении интервала дозирования в равновесном состоянии (AUCt,ss). Вторичными параметрами являются Cmax и Ctrough в равновесном состоянии.
и, как правило, Cmax - при подкожном. Необходимо также оценить такие вторичные параметры, как tmax, объем распределения и период полувыведения. Основными параметрами в исследовании при многократном введении являются AUC, усеченная с момента первого введения до второго введения (AUC0-t), и AUC на протяжении интервала дозирования в равновесном состоянии (AUCt,ss). Вторичными параметрами являются Cmax и Ctrough в равновесном состоянии.
В любом ФК-исследовании наряду с ФК-оценкой необходимо определять антитела к препарату, используя соответствующие временные точки взятия образцов.
5.2. Фармакодинамические исследования
К исследованиям фармакокинетики рекомендуется добавлять определение фармакодинамических маркеров, если это возможно. Фармакодинамические маркеры должны быть отобраны на основании их клинической значимости.
В некоторых случаях может оказаться достаточным подтверждения клинической сопоставимости биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата с помощью сравнительных ФК-исследований (ФД-исследований) при выполнении следующих условий.
Выбранный ФД-маркер (биомаркер) является принятым суррогатным маркером и соотносится с исходом у пациентов в такой степени, что подтверждение аналогичного влияния на ФД-маркер будет обеспечивать аналогичное влияние на клинический исход. К подходящим примерам относятся абсолютное число нейтрофилов при оценке влияния гранулоцитарного колониестимулирующего фактора (Г-КСФ), снижение ранней вирусной нагрузки при хроническом гепатите C при оценке влияния альфа-интерферонов и эугликемический клэмп-тест - в целях сравнения двух инсулинов. Для сравнения двух 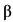 -интерферонов можно прибегнуть к магнитно-резонансной томографии очагов при рассеянном склерозе.
-интерферонов можно прибегнуть к магнитно-резонансной томографии очагов при рассеянном склерозе.
Некоторые ФД-маркеры не являются подтвержденными суррогатами эффективности, но значимы для фармакологического действия фармацевтической субстанции и имеют четкую зависимость доза - эффект или концентрация - эффект. В этом случае во избежание исследования клинической эффективности может оказаться достаточным исследования доза - экспозиция - ответ при однократном или многократном введении двух или более доз. Такой дизайн обеспечит сравнение биоаналога и препарата сравнения в пределах крутой части кривой доза-эффект (аналитическая чувствительность, в соответствии с актами, входящими в право Союза).
В исключительных случаях подтверждающее клиническое исследование может не потребоваться при условии надежного подтверждения сопоставимости в рамках оценки биоаналогичности (биоподобия) с помощью физико-химических, структурных и in vitro биологических испытаний и ФК-исследований у человека вместе с комбинацией ФД-маркеров, отражающих фармакологическое действие и концентрацию активной фармацевтической субстанции.
Если подтверждение сопоставимости клинической эффективности и безопасности в рамках оценки биоаналогичности (биоподобия) основано на ФК-исследованиях, подкрепленных исследованиями с несуррогатными ФД-маркерами (биомаркерами), подобный подход ("отпечатки пальцев") рекомендуется обсудить с уполномоченными органами. План должен включать величину границ эквивалентности с ее клиническим обоснованием, а также меры по подтверждению сопоставимого профиля безопасности.
5.3. Исследования эффективности
В отсутствие суррогатных маркеров эффективности, как правило, требуется подтверждение сопоставимой клинической эффективности биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата в рандомизированном параллельном сравнительном исследовании с достаточной мощностью, предпочтительно, двойном слепом с использованием конечных точек эффективности. Исследуемая популяция должна отражать зарегистрированное показание к применению оригинального (референтного) лекарственного препарата и быть чувствительной для выявления потенциальных различий между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом. Изменение клинической практики может потребовать отклонения от зарегистрированного показания к применению, например, в части сопутствующей терапии, используемой в составе комбинированной терапии, очередности назначения препаратов или тяжести заболевания. Отклонения необходимо обосновать и обсудить с уполномоченными органами.
5.3.1. Дизайны исследований.
Рекомендуется использовать дизайн эквивалентности. Использование дизайна не меньшей эффективности приемлемо, если оно обосновано строгими научными данными и с учетом характеристик оригинального (референтного) лекарственного препарата, например, профиля безопасности и переносимости, диапазона доз, зависимости доза - эффект. Исследование не меньшей эффективности допустимо, если, исходя из научных и механистических оснований, можно исключить возможность существенного и клинически значимого повышения эффективности. Однако, как и в исследованиях эффективности, необходимо учесть аналитическую чувствительность.
Дизайн не меньшей эффективности рекомендуется обсудить с уполномоченными органами.
5.3.2. Конечные точки эффективности.
Исследования эффективности биоаналогичных (биоподобных) лекарственных препаратов не нацелены на подтверждение эффективности per se, поскольку она ранее уже была подтверждена для оригинального (референтного) лекарственного препарата. Целью исследований эффективности является подтверждение сопоставимости клинических характеристик биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата.
Составлены руководства по заболеваниям для разработки инновационных лекарственных препаратов. При разработке биоаналогичного (биоподобного) лекарственного препарата выбор клинических конечных точек и сроков проведения анализа конечных точек может не совпадать с рекомендациями в отношении новых действующих веществ. Настоящими Правилами предусмотрены специфичные для классов препаратов требования, чтобы направлять разработку биоаналогичных (биоподобных) лекарственных препаратов в определенных областях. При отсутствии в настоящих Правилах соответствующих требований, сопоставимость следует подтверждать с помощью достаточно чувствительных клинических моделей и условий исследования. Заявитель должен обосновать, что выбранная модель релевантна и чувствительна для выявления потенциальных различий в части эффективности и безопасности. Тем не менее отклонение от рекомендуемых руководствами по заболеваниям конечных точек требует научного обоснования. Обнаруженные различия в эффективности между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом всегда требуют анализа их клинической значимости. В целом цель клинических данных заключается в оценке небольших различий, обнаруженных на предыдущих этапах, и подтверждении аналогичности клинических характеристик биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата. Использовать клинические данные с целью обоснования существенных различий в показателях качества не допускается.
Корреляция между "твердыми" клиническими конечными точками, рекомендуемыми руководствами в отношении новых действующих веществ, и другими клиническими и фармакодинамическими конечными точками, которые более чувствительны для выявления клинических значимых различий, может быть подтверждена с помощью ранее проведенных клинических исследований оригинального (референтного) лекарственного препарата. В этом случае необязательно использовать те же первичные конечные точки эффективности, которые были использованы для регистрации оригинального (референтного) лекарственного препарата. Рекомендуется включить некоторые общие конечные точки (например, в качестве вторичных конечных точек), в целях содействия сравнению клинических исследований, проведенных с референтным лекарственным препаратом.
Необходимо заранее оговорить границы сопоставимости и обосновать их статистически и клинически, используя данные об оригинальном (референтном) лекарственном препарате в соответствии с актами, входящими в право Союза. Подобно всем клиническим исследованиям со сравнительным дизайном, необходимо учесть аналитическую чувствительность в соответствии с актами, входящими в право Союза.
5.4. Клиническая безопасность
Клиническая безопасность важна на протяжении всей программы клинической разработки и определяется в ходе начальных ФК-исследований и (или) ФД-исследований, а также в составе опорных клинических исследований эффективности. Данные о сравнительной безопасности следует (в норме) собирать на предрегистрационном этапе, их количество зависит от вида и тяжести нарушений, вызываемых оригинальным (референтным) лекарственным препаратом. Необходимо обосновать продолжительность наблюдения за безопасностью на предрегистрационном этапе. Необходимо тщательно оценивать разновидность, тяжесть и частоту нежелательных реакций между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом, особенно тех, которые описаны в общей характеристике лекарственного препарата последнего. В регистрационном досье заявитель должен представить оценку конкретных рисков, ожидаемых в отношении биоаналогичного (биоподобного) лекарственного препарата. Она, в частности, включает описание возможных опасений со стороны безопасности, которые могут быть обусловлены процессом производства, отличающимся от такового оригинального (референтного) лекарственного препарата, в особенности риски инфузионных реакций и иммуногенности.
Принципы оценки иммуногенности терапевтических белков и моноклональных антител описаны в главах 11 и 12 настоящих Правил. Иммуногенный потенциал биоаналогичного (биоподобного) лекарственного препарата необходимо сравнить с таковым оригинального (референтного) лекарственного препарата, следуя принципам, изложенным в указанных главах, если только не представлено обоснование необходимости отклонения от этого подхода. Вид и объем данных об иммуногенности зависят от опыта применения оригинального (референтного) лекарственного препарата и класса препарата.
Испытание иммуногенности биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата следует проводить в процессе исследований сопоставимости в рамках оценки биоаналогичности (биоподобия), используя формат испытаний и схему взятия образцов, которые должны удовлетворять всем действующим стандартам. Аналитические испытания необходимо проводить как с препаратом сравнения, так и с молекулой биоаналогичного (биоподобного) лекарственного препарата параллельно (с ослеплением), чтобы измерить иммунный ответ на препарат, который был получен каждым пациентом. Предпочтительно, чтобы аналитические испытания были способны обнаруживать антитела как к биоподобному лекарственному препарату, так и к молекуле оригинального (референтного) лекарственного препарата, но по меньшей мере обладали способностью обнаруживать все антитела, выработанные на молекулу биоаналогичного (биоподобного) лекарственного препарата. Измерять и представлять, как правило, следует частоту возникновения и свойства (например, перекрестную реактивность, эпитопы-мишени и нейтрализующую активность) антител и титры антител, а также оценивать и интерпретировать их во взаимосвязи с потенциальным влиянием на параметры клинической эффективности и безопасности.
Продолжительность исследования иммуногенности следует обосновывать в индивидуальном порядке, исходя из продолжительности курса терапии, выведения препарата из кровотока (во избежание влияния антигена на методики) и сроков формирования гуморального иммунного ответа (по меньшей мере через четыре недели при применении иммунодепрессанта). Продолжительность последующего наблюдения следует обосновывать сроками возникновения и характеристиками нежелательного иммунного ответа, описанными для оригинального (референтного) лекарственного препарата, например, низким риском клинически значимой иммуногенности или несущественным трендом повышения иммуногенности во времени. При хроническом применении на предрегистрационном этапе, как правило, требуется представить данные годичного наблюдения. Данные наблюдения за более короткий срок (например, 6 месяцев) могут быть обоснованы, исходя из профиля иммуногенности оригинального (референтного) лекарственного препарата. При необходимости на пострегистрационном этапе могут впоследствии потребоваться дополнительные данные об иммуногенности за период до одного года. Рекомендации к отдельным препаратам представлены в главах 15.3 - 15.11.
Повышенная по отношению к оригинальному (референтному) лекарственному препарату иммуногенность может затруднить анализ отношения пользы к рискам и поставить биоаналогичность под сомнение. Однако биоаналогичный (биоподобный) лекарственный препарат может обладать и меньшей иммуногенностью, что не будет препятствием для регистрации его в качестве биоаналогичного (биоподобного) лекарственного препарата. Если на биоаналогичный (биоподобный) лекарственный препарат образуется меньше нейтрализующих антител, анализ эффективности всей исследуемой популяции может привести к ошибочному заключению, что биоаналогичный (биоподобный) лекарственный препарат эффективнее оригинального (референтного) лекарственного препарата. В связи с этим рекомендуется заранее предусмотреть дополнительную поисковую подгруппу с целью анализа эффективности и безопасности у тех пациентов, у которых в ходе клинического исследования антитела к препарату не образовывались. Такой анализ может способствовать установлению того, что эффективность биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) лекарственного препарата аналогична, если исключить влияние иммунного ответа.
6. Экстраполяция эффективности и безопасности
с одного показания к применению на другое
Оригинальный (референтный) лекарственный препарат может иметь несколько показаний к применению. Если сопоставимость в рамках оценки биоаналогичности (биоподобия) была подтверждена в отношении одного из них, возможна экстраполяция клинических данных на другие показания к применению оригинального (референтного) лекарственного препарата, но она требует научного обоснования. Если отсутствует определенность в отношении того, что безопасность и эффективность, подтвержденные в отношении одного показания к применению, будут релевантны для другого, потребуются дополнительные данные. Экстраполяцию следует проводить в совокупности всех данных, то есть данных по качеству, доклинических и клинических данных. Предполагается, что экстраполяция безопасности и эффективности возможна, когда сопоставимость в рамках оценки биоаналогичности (биоподобия) была подтверждена с помощью доскональных физико-химических и структурных анализов, а также функциональных испытаний in vitro, подкрепленных клиническими данными (эффективности и безопасности и (или) ФК-данными (ФД-данными)). В определенных ситуациях требуются дополнительные данные, например:
действующее вещество оригинального (референтного) лекарственного препарата взаимодействует с несколькими рецепторами, которые могут оказывать различное влияние при изученных и не изученных показаниях к применению;
само действующее вещество имеет несколько активных центров, которые могут оказывать различное влияние при различных показаниях к применению;
изученное показание к применению не релевантно для других с позиций эффективности или безопасности, т.е. не обладает чувствительностью в отношении различий во всех значимых аспектах эффективности и безопасности.
Иммуногенность опосредована множеством факторов, включая путь введения, режим дозирования, факторы, опосредованные пациентами, и факторы, опосредованные заболеванием (например, сопутствующая терапия, разновидность заболевания, иммунный статус). Таким образом, иммуногенность при различных показаниях может различаться. Экстраполяция иммуногенности с изученного показания или пути введения на другие требует обоснования.
7. Фармаконадзор
Для выявления редких нежелательных реакций данных предрегистрационных клинических исследований, как правило, недостаточно. Следовательно, необходимо на постоянной основе вести пристальное наблюдение за клинической безопасностью биоаналогичных (биоподобных) лекарственных препаратов в пострегистрационную фазу, включая непрерывную оценку пользы и рисков.
В рамках процедуры регистрации заявитель должен представить описание системы фармаконадзора и план управления рисками в соответствии с актами, входящими в право Союза, включая правила надлежащей практики фармаконадзора Союза, утверждаемые Комиссией. План управления рисками должен учитывать выявленные и потенциальные риски, присущие применению оригинального (референтного) лекарственного препарата, с описанием их учета в ходе пострегистрационного наблюдения. В этой связи необходимо отдельно рассмотреть иммуногенность.
В плане фармаконадзора биоаналогичного (биоподобного) лекарственного препарата необходимо должным образом отразить любой особый мониторинг безопасности, требуемый в отношении оригинального (референтного) лекарственного препарата или класса препаратов. Заявителям рекомендуется принимать участие во всех проводимых фармакоэпидемиологических исследованиях оригинального (референтного) лекарственного препарата. Однако может потребоваться проведение новых исследований. Действия по минимизации рисков, принимаемые в отношении оригинального (референтного) лекарственного препарата, следует в принципе также включать в программу управления рисками биоаналогичного (биоподобного) лекарственного препарата. Любые отклонения от указанного выше требуют обоснования (например, если минимизация рисков обусловлена изделием, используемым вместе с препаратом сравнения).
В отношении подозреваемых нежелательных реакций, обусловленных биологическими лекарственными препаратами, особую важность представляет точная идентификация рассматриваемого препарата с точки зрения его производства. Исходя из этого, следует принять все необходимые меры по четкой идентификации биологического лекарственного препарата, являющегося объектом сообщения о подозреваемой нежелательной реакции, с точным указанием его торгового наименования и номера серии.
Глава 15.3. Биоаналогичные (биоподобные) лекарственные
препараты, содержащие моноклональные антитела. Вопросы
доклинических и клинических исследований
1. Область применения
Настоящая глава разработана в отношении моноклональных антител, она дополняет главу 15.2 настоящих Правил и содержит требования для подтверждения сопоставимости двух лекарственных препаратов, содержащих моноклональные антитела (МкАТ), с целью составления регистрационного досье. Несмотря на то, что настоящая глава составлена специально для моноклональных антител, обсуждаемые в ней принципы применимы к родственным веществам, например, к гибридным белкам на основе Fc-фрагмента IgG(-цепт молекулы).
МкАТ нового поколения, то есть МкАТ с измененной по отношению к зарегистрированным оригинальным (референтным) лекарственным препаратам структурой и (или) функциональностью (например, глико-инженерные МкАТ с повышенной активностью), направленной на улучшение или изменение клинических эффектов, не являются биоаналогичными (биоподобными) лекарственными препаратами и поэтому в настоящей главе не рассматриваются.
В настоящей главе приведены указания по доклиническим и клиническим исследованиям лекарственных препаратов, содержащих МкАТ, заявленных в качестве биоаналогичных (биоподобных) лекарственных препаратов по отношению к зарегистрированному биологическому лекарственному препарату МкАТ. В разделе по доклиническим исследованиям описаны фармако-токсикологические требования. В разделе по клиническим исследованиям представлены требования к исследованиям сравнительной фармакокинетики, фармакодинамики, эффективности и безопасности, а также фармаконадзору.
Основная цель настоящей главы - установить общие принципы, которые позволят заявителю разработать программу, на основании которой можно установить сопоставимость биоаналогичного (биоподобного) МкАТ с оригинальным (референтным) МкАТ, сохраняя при этом ранее доказанную безопасность и эффективность препарата. В ходе разработки программы рекомендуется придерживаться пошагового подхода: объем и характер доклинических и клинических исследований зависят от результатов, полученных на предыдущем этапе. Все исследования должны быть направлены на выявление потенциальных различий между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом, а также при возникновении таких различий на установление их значимости.
В целях индивидуального подхода к выбору и объему исследований in vitro и in vivo в ходе доклинической разработки рекомендуется придерживаться пошагового подхода к изучению МкАТ. Сначала для анализа функциональных различий и различий в связывании (с рецептором) необходимо провести сравнительные исследования in vitro. На втором этапе следует определить необходимость проведения дополнительных доклинических исследований in vivo. При необходимости проведения исследования in vivo его цель будет зависеть от требуемых дополнительных сведений и наличия релевантной животной модели. Проведение токсикологических исследований на нечеловекообразных приматах не рекомендуется.
В ходе программы клинической разработки число вовлеченных пациентов, как правило, должно соответствовать (быть соразмерным) уровню доказательств, полученных на предыдущих этапах, которые подтверждают сопоставимость (подобие) лекарственного препарата. Начальным этапом разработки биоаналогичного (биоподобного) МкАТ, как правило, является сравнительное фармакокинетическое исследование у достаточно чувствительной и однородной популяции исследования (здоровые добровольцы или пациенты). Фармакокинетические данные могут помочь при решении вопроса об экстраполяции данных по безопасности и эффективности между различными показаниями оригинального (референтного) МкАТ. В некоторых случаях, устанавливаемых в индивидуальном порядке, может потребоваться проведение нескольких фармакокинетических исследований с многократным введением препарата у пациентов или потребоваться введение фармакокинетического этапа в клиническое исследование, направленное на подтверждение аналогичной безопасности и эффективности. По возможности фармакокинетические исследования допускается комбинировать с фармакодинамическими конечными точками. Сопоставимую клиническую эффективность необходимо подтвердить, как правило, с помощью достаточно мощного рандомизированного параллельного сравнительного клинического исследования эквивалентности, предпочтительно двойного слепого. В целях подтверждения сопоставимости может потребоваться отклониться от руководств, составленных по отдельным заболеваниям. Основополагающий принцип заключается в подтверждении аналогичной клинической эффективности и безопасности по отношению к оригинальному (референтному) лекарственному препарату, а не в установлении пользы для пациента per se, которая для оригинального (референтного) лекарственного препарата ранее уже была доказана. Следует использовать наиболее чувствительные модели и условия исследования (фармакодинамического или клинического) у однородной группы пациентов. Если для подтверждения аналогичной эффективности наиболее целесообразными являются сравнительные фармакодинамические исследования, заявители должны подобрать клинически значимые маркеры, обосновать свой выбор, а также представить достаточные данные по клинической безопасности, особенно иммуногенности. Основываясь на совокупных имеющихся результатах исследований сопоставимости и при должном обосновании, допускается экстраполяция данных клинической эффективности и безопасности на прочие показания к применению оригинального (референтного) МкАТ, которые в ходе клинической разработки отдельно не изучались. Концепция пострегистрационного наблюдения, предлагаемая заявителями, может превосходить стандартные требования к фармаконадзору и включать пострегистрационные исследования безопасности.
МкАТ являются основным классом лекарственных препаратов, получаемых биотехнологическим путем. Различные препараты МкАТ обладают некоторыми общими свойствами, например, цитотоксичностью в отношении их мишени или нейтрализацией цитокина, но различаются по таким свойствам, как механизм действия. МкАТ имеют сложную структуру и в зависимости от изотипа могут содержать несколько функциональных доменов в молекуле (антигенсвязывающий участок, комплементсвязывающий участок, константная часть, взаимодействующая с Fc-рецепторами). Каждое МкАТ имеет уникальный профиль с точки зрения антигенсвязывающего участка, эффекторной функции Fc-цитотоксичности и связывания с Fc-рецепторами. В последние годы разработано множество методов подробного установления характеристик сложных белков как на физико-химическом, так и на функциональном уровне, например, методы количественного определения активности; накоплен опыт оценки незначительных различий между показателями качества, обусловленными изменениями процесса производства МкАТ. Однако современный уровень знаний не позволяет интерпретировать значимость небольших различий между физико-химическими и биологическими характеристиками, обнаруженными при сопоставлении биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ.
2. Сфера применения
Настоящая препаратспецифичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих МкАТ.
3. Связь с другими главами
В главах 15 - 15.2 содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Доклинические исследования
При оценке аналогичности биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ в доклинической разработке применяется пошаговый подход.
Доклинические исследования должны быть проведены перед началом клинических исследований. Сначала необходимо провести исследования in vitro, а затем определить необходимость и объем проведения требуемых исследований in vivo.
В доклиническом обзоре регистрационного досье (модуль 2.4) необходимо полностью обосновать выбранный подход.
4.1. Шаг 1. Исследования в условиях in vitro
Чтобы оценить различия в биологической активности между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами, необходимо представить данные ряда сравнительных исследований в условиях in vitro, некоторые из которых могут быть уже доступны по результатам исследований качества.
Доклинические исследования in vitro необходимо проводить, используя достаточное число серий препарата, отражающих свойства серий, которые будут использоваться в клинических исследованиях. Такие исследования должны включать установление:
связывания с антигеном-мишенью (антигенами-мишенями);
связывания с репрезентативными изоформами соответствующих трех  -рецепторов (
-рецепторов (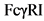 ,
, 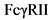 и
и 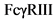 ), FcRn и комплементом (C1q);
), FcRn и комплементом (C1q);
Fab-ассоциированных функций (например, нейтрализации растворимого лиганда, активации или блокады рецептора);
Fc-ассоциированных функций (например, антителозависимой клеточной цитотоксичности (АЗКЦ), комплементзависимой цитотоксичности (КЗЦ), активации комплемента).
Указанные исследования должны носить сравнительный характер и быть достаточно чувствительными, позволяющими обнаружить различия в зависимости "концентрация - активность" между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом, и не должны проводиться исключительно с целью изучения этих свойств per se. Следует отметить, что для МкАТ, направленных на немембранные мишени, изучать АЗКЦ и КЗЦ не требуется. Исследования перекрестной тканевой реактивности не пригодны для обнаружения незначительных изменений ключевых параметров качества и, таким образом, не рекомендуются для изучения сопоставимости.
Такие исследования должны в совокупности широко охватывать функциональные свойства МкАТ, несмотря на то, что некоторые из них могут быть незначимы для реализации терапевтического эффекта. Поскольку исследования in vitro могут оказаться более специфичными и чувствительными, чем исследования на животных, они могут играть основную роль в подтверждении доклинической сопоставимости.
Если по результатам изучения сопоставимости с помощью описанной выше стратегии обнаруживается, что биоаналогичное (биоподобное) МкАТ и оригинальное (референтное) МкАТ нельзя признать биоаналогичными (биоподобными), следует рассмотреть возможность разработки препарата как самостоятельного.
4.2. Шаг 2. Установление потребности в исследованиях
в условиях in vivo
Общепризнано, что некоторые МкАТ могут опосредовать эффекты, которые невозможно полностью выявить с помощью исследований in vitro. В связи с этим могут потребоваться исследования in vivo при условии, что имеется релевантная по виду животного и дизайну модель in vivo. При определении необходимости проведения дополнительных доклинических исследований in vivo необходимо рассмотреть ряд факторов (перечень не исчерпывающий):
наличие значимых показателей качества, которые не были обнаружены в оригинальном (референтном) лекарственном препарате (например, новая посттрансляционная структурная модификация);
наличие показателей качества, значимо различающихся количественно от таковых оригинального (референтного) лекарственного препарата;
значимые различия по составу, например, наличие вспомогательных веществ, редко используемых в препаратах МкАТ.
Несмотря на то, что каждый из упомянутых факторов не обязательно требует испытаний in vivo, для определения степени настороженности и необходимости проведения испытаний in vivo их следует рассматривать одновременно.
Если результаты исследований сопоставимости in vitro, проведенные на шаге 1, признаются удовлетворительными, а на шаге 2 факторы настороженности не обнаруживаются или такие факторы настороженности не препятствуют прямому применению у человека, исследование на животных in vivo допускается не проводить.
При необходимости дополнительных сведений следует учитывать наличие подходящих животных моделей или иных подходящих моделей (например, трансгенных животных или трансплантатов). Ввиду специфичности МкАТ релевантными для изучения животными моделями в большинстве случаев являются нечеловекообразные приматы. Во всех случаях необходимо принимать во внимание ограничения исследования in vivo (например, чувствительность и вариабельность).
В отсутствие релевантной животной модели in vivo заявитель вправе начать исследования у человека, принимая во внимание принципы по снижению потенциальных рисков.
4.3. Шаг 3: Исследования в условиях in vivo
При необходимости проведения исследования in vivo их направленность (ФК, и (или) ФД, и (или) безопасность) определяется требуемыми сведениями. Понятие "безопасностью" в данном случае означает не полное исследование токсичности при многократном введении, а, скорее, прижизненный анализ параметров безопасности, таких как клинические признаки, масса тела и жизненно важные функции. Исследования на животных необходимо спланировать таким образом, чтобы получить максимальный объем информации. В зависимости от оцениваемых конечных точек не всегда существует необходимость заканчивать исследование гибелью животных. При планировании исследования in vivo необходимо руководствоваться принципом 3R (reduce - refine - replace, сокращение - улучшение - замена). Приняв во внимание фармакокинетические свойства МкАТ и их клиническое применение, необходимо обосновать продолжительность исследования (включая период наблюдения).
Если позволяет модель, ФК и ФД биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов необходимо сравнить количественно, в том числе провести анализ "концентрация - эффект", охватывающий терапевтические дозы у человека.
Проведение токсикологических исследований на нечеловекообразных приматах, как правило, не рекомендуется. К тому же не рекомендуется проводить токсикологические исследования на нерелевантных видах животных (например, изучать исключительно неспецифическую токсичность, обусловленную примесями). В силу различий в процессах производства биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов могут возникать качественные различия в профилях производственных примесей (например, белках клетки-хозяина). Необходимо обеспечить наименьшее содержание таких примесей, что является наилучшей стратегией минимизации обусловленных ими рисков. Качественные или количественные различия в родственных вариантах (например, профили гликозилирования, варианты с различным зарядом могут повлиять на биологические функции МкАТ, поэтому их необходимо изучить с помощью подходящих in vitro методов количественного определения. Такие различия по качеству могут оказать влияние на иммуногенный потенциал или возможность развития гиперчувствительности. Общепризнано, что указанные эффекты с помощью исследований на животных сложно спрогнозировать, поэтому они требуют дальнейшего изучения в клинических исследованиях. Оценка иммуногенности на животных в целом обладает низкой прогностической значимостью для иммуногенности у человека, однако такие данные могут потребоваться для интерпретации результатов исследований на животных in vivo. В целях будущих исследований (при возникновении потребности в них) необходимо отобрать и обеспечить хранение образцов крови.
При доклиническом подтверждении биоаналогичности МкАТ исследования фармакологической безопасности и репродуктивной токсичности не требуются. Исследования местной переносимости, как правило, не требуются. Если вводятся вспомогательные вещества, в отношении которых отсутствует опыт их клинического применения при заявленном пути введения или такой опыт сильно ограничен, могут потребоваться исследования местной переносимости. Чтобы не проводить отдельные исследования местной переносимости, при проведении прочих исследований in vivo изучение местной переносимости может являться их частью.
5. Клинические исследования
Сравнительные клинические исследования биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов необходимо проводить во всех случаях. Количество и вид исследований зависят от оригинального (референтного) лекарственного препарата, их необходимо строго научно обосновать. В программе разработки рекомендуется, как правило, придерживаться пошагового подхода, объем и характер клинической программы зависят от результатов, полученных на предыдущем этапе. В ходе программы клинической разработки число вовлеченных пациентов, как правило, должно соответствовать (быть соразмерным) уровню доказательств, полученных на предыдущих этапах, которые подтверждают сопоставимость (подобие) лекарственного препарата.
5.1. Шаг 1. Исследование фармакокинетических свойств
Первым этапом разработки биоаналогичного (биоподобного) МкАТ, как правило, является сопоставление фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов. Дизайн исследования зависит от ряда факторов, включая клинические особенности, безопасность, фармакокинетическую характеристику антитела (мишень-опосредованная диспозиция (связывание, распределение, метаболизм и элиминация), линейная или нелинейная ФК, временная зависимость, период полувыведения и др.), и должен учитывать рекомендации, изложенные в правилах проведения исследований биоэквивалентности лекарственных препаратов в рамках Союза и иных актах, входящих в право Союза. Более того, биоаналитические методики должны соответствовать их целевому назначению и требуют должной валидации в соответствии с правилами проведения исследований биоэквивалентности лекарственных препаратов в рамках Союза.
5.1.1. Дизайн (программа) исследования.
Основной целью фармакокинетических исследований, результаты которых представляются в регистрационном досье биоаналогичного (биоподобного) лекарственного препарата, является сопоставимость фармакокинетики биоаналогичного (биоподобного) лекарственного препарата с фармакокинетикой оригинального (референтного) лекарственного препарата у достаточно чувствительной и однородной популяции. Считается, что в этом случае снижается вариабельность и, следовательно, размер выборки, необходимый для подтверждения эквивалентности, что позволяет облегчить интерпретацию результатов.
У здоровых добровольцев может отмечаться более низкая фармакокинетическая вариабельность, поскольку по сравнению с пациентами мишень-опосредованный клиренс у них играет меньшую роль. В связи с этим (по возможности) для получения важных сведений о биоаналогичности рекомендуется провести исследование с однократным введением препарата у здоровых добровольцев. С фармакокинетической точки зрения в целях всесторонней характеристики ФК-профиля, включая фазу поздней элиминации, предпочтительно провести перекрестное исследование с однократным введением. Ввиду длительного периода полувыведения МкАТ и потенциального влияния на иммуногенность может потребоваться проведение исследования с параллельным дизайном.
При токсическом механизме действия или при недостаточности сведений для установления биоаналогичности исследование у здоровых добровольцев может быть нецелесообразным. В этом случае предпочтительно провести исследование у пациентов. Если исследование с однократным введением у пациентов нецелесообразно, проводят исследование с многократным введением.
Может возникнуть необходимость проведения ФК-исследования у популяции, отличной от той, у которой будет подтверждаться аналогичность по клинической эффективности, поскольку наиболее чувствительная популяция для сравнения ФК-характеристик может отличаться от популяции, в которой наиболее целесообразно подтверждать аналогичность эффективности и безопасности. В этом случае в рамках исследования клинической эффективности рекомендуется определять популяционные ФК-параметры, поскольку такие данные могут дополнить совокупную базу данных по подтверждению сопоставимости.
Основываясь на всестороннем изучении научной литературы в части чувствительности исследования и возможности распространения ФК-результатов на прочие клинические показания, утвержденные для оригинального (референтного) МкАТ, необходимо полностью обосновать выбор популяции для ФК-исследования.
Если ФК-исследование у здоровых добровольцев проводится с целью дополнительного подтверждения биоэквивалентности, в рамках клинических исследований у пациентов рекомендуется осуществлять сбор вспомогательных ФК-данных, которые могут служить веским обоснованием аналогичности ФК-свойств.
На подготовку плана ФК-анализа могут оказать влияние следующие факторы.
Характеристики заболевания и пациентов. К факторам, которые могут повлиять на выбор популяции пациентов, относятся: возраст типичной манифестации заболевания и возрастной диапазон (поскольку в более молодом возрасте вероятность сопутствующей патологии ниже), объем предыдущего лечения, сопутствующая терапия и уровень экспрессии антигена (которая может зависеть от стадии заболевания). В отношении МкАТ, применяющихся как в монотерапии, так и в составе комбинированной терапии с иммунодепрессантами или химиотерапии, в целях снижения источников вариабельности целесообразно проводить сравнительное ФК-исследование в условиях монотерапии. Однако в некоторых случаях целесообразно включать в популяцию пациентов, получающих первую линию терапии или адъювантную терапию на ранних этапах рака при низкой опухолевой нагрузке. В таких случаях МкАТ, как правило, вводят в составе комбинированной терапии.
Фармакокинетические характеристики оригинального (референтного) МкАТ. Фармакокинетика противоопухолевых МкАТ может носить зависимый от времени характер, поскольку после многократного введения опухолевая нагрузка может измениться (например, вследствие увеличения периода полувыведения при многократном введении), что требует учета при планировании исследования.
Наличие двух механизмов клиренса (зависимого и не зависимого от мишени может повлиять на количество необходимых исследований. Если мишень-опосредованный клиренс незначим, как правило, достаточно одного ФК-исследования. Если оригинальное (референтное) МкАТ элиминируется с помощью как мишень-опосредованного, так и неопосредованного механизмов, необходимо подтвердить сопоставимость фармакокинетики, при которой каждый из механизмов клиренса преобладает: рекомендуется провести одно исследование у здоровых добровольцев на предмет мишень-неопосредованного клиренса и одно вспомогательное исследование у пациентов, которое может являться частью исследования эффективности и быть направлено на установление сопоставимости по мишень-опосредованному клиренсу.
В отношении мишеней МкАТ, включающих рецепторы, экспрессируемые на опухолевых клетках и которые подвергаются шеддингу, в целях установления исходной сопоставимости сравниваемых групп рекомендуется измерять содержание сброшенных рецепторов до начала и при необходимости в ходе проведения исследования. Стратификация по опухолевой нагрузке или сбросу рецепторов (если таковая возможна) позволяет удостовериться в исходной сопоставимости. Целесообразно провести поисковый статистический анализ последующей сопоставимости во временных точках, значимых для составления заключения о ФК-эквивалентности.
Для зарегистрированных по нескольким показаниям МкАТ определять фармакокинетический профиль по каждому из них, как правило, не требуется. Однако, если препарат МкАТ используется в различных областях медицины (например, иммунологии и онкологии), могут потребоваться отдельные ФК-исследования, поскольку в различных областях мишень-опосредованный клиренс может различаться.
Дозы. Фактически тестировать все терапевтические схемы приема препарата (указанные в инструкции по применению) не требуется. Необходимо выбрать наиболее чувствительную дозировку, чтобы выявить потенциальные различия фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов. При наличии ограниченных данных о том, какая дозировка является наиболее чувствительной, рекомендуется исследовать низкую или минимальную рекомендуемую терапевтическую дозу, при которой подразумевается, что мишень-опосредованный клиренс еще не достиг максимума, и высокую или максимальную терапевтическую дозу, при которой подразумевается, что доминирует неспецифический механизм клиренса. Исследование однократного введения с минимальной терапевтической дозировкой у пациентов считается наиболее приемлемым для исследования различий мишень-опосредованного клиренса (при наличии).
Пути введения. Если оригинальный (референтный) лекарственный препарат вводится и внутривенно, и подкожно, а биоаналогичный (биоподобный) лекарственный препарат предполагается вводить обоими путями, рекомендуется изучить оба пути введения. Однако поскольку анализ подкожного пути введения охватывает как абсорбцию, так и элиминацию, можно отказаться от изучения внутривенного пути введения, если при использовании дополнительных ФК-параметров, например, частичных AUC (в соответствии с разделом 3.1.2), в отношении подкожного пути введения показана сопоставимость и по абсорбции, и по элиминации.
5.1.2. Время отбора проб. В исследованиях с однократным введением режим отбора проб должен охватывать весь профиль, включая фазу поздней элиминации. Для лекарственных препаратов, вводящихся 2 и более раза, ценными являются сведения, полученные как после первого, так и после последнего введения, поскольку первое введение предпочтительно в сравнительных целях, а последнее дает информацию о фазе терминальной элиминации, которую невозможно получить после первого введения.
Если в целях подтверждения аналогичности между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом используется фармакокинетическое исследование с многократным введением препаратов пациентам и если элиминацию после введения последней дозы охарактеризовать не удается, отбор проб должен позволить охарактеризовать профиль "концентрация - время" как после введения первой дозы, так и после введения последующих (предпочтительно в равновесном состоянии). Установление характеристик полного профиля "концентрация - время" в равновесном состоянии наиболее целесообразно при нелинейной фармакокинетике оригинального (референтного) МкАТ (например, многие противоопухолевые МкАТ с клеточными мишенями проявляют дозозависимую и зависимую от времени ФК или затрагивающие иммуногенность изменения в кинетике деградации или элиминации).
5.1.3. Исследуемые фармакокинетические параметры. Первичным параметром в исследовании с однократным введением должна стать 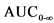 . Необходимо также изучить вторичные параметры, такие как Cmax, tmax, объем распределения и период полувыведения. При подкожном введении дополнительным первичным параметром становится Cmax. Кроме того, если какие-либо данные о внутривенном пути отсутствуют, для обеспечения сопоставимости как в фазу абсорбции, так элиминации необходимо изучить частичные AUC.
. Необходимо также изучить вторичные параметры, такие как Cmax, tmax, объем распределения и период полувыведения. При подкожном введении дополнительным первичным параметром становится Cmax. Кроме того, если какие-либо данные о внутривенном пути отсутствуют, для обеспечения сопоставимости как в фазу абсорбции, так элиминации необходимо изучить частичные AUC.
Первичным параметром в исследовании с многократным введением должна стать AUC после первого введения, усеченная до второго введения (AUC0-t), и AUC за интервал дозирования в равновесном состоянии 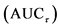 . Вторичные параметры: Cmax и Cmin в равновесном состоянии.
. Вторичные параметры: Cmax и Cmin в равновесном состоянии.
Используя наиболее подходящие временные точки, параллельно с изучением ФК необходимо определять антитела к препарату.
Необходимо заранее указать и обосновать границы сопоставимости. Известно о высокой межиндивидуальной вариабельности некоторых оригинальных (референтных) МкАТ по некоторым параметрам, что необходимо учитывать при выборе границ сопоставимости по крайней мере этих параметров. Всякое расширение стандартной границы эквивалентности за пределы 80 - 125 процентов для первичных параметров требует подробного обоснования, включая оценку потенциального влияния на клиническую эффективность и безопасность. В отношении вторичных параметров достаточно указать доверительные интервалы для отношения или разности и описательные статистики, диапазоны допустимости определять не требуется. Необходимо проанализировать клиническую значимость полученных различий и соответствующих им доверительных интервалов.
5.1.4. Сроки ФК-анализа.
Исследованиям клинической эффективности должно, как правило, предшествовать подтверждение аналогичности ФК-профилей. Однако в некоторых случаях, например, при неизбежно высокой ФК-вариабельности МкАТ даже по одному показанию к применению, из практических соображений сравнительный ФК-анализ целесообразно провести в рамках клинического исследования, направленного на подтверждение аналогичной клинической эффективности (поскольку лишь такое исследование будет достаточно крупным для подтверждения ФК-эквивалентности). В зависимости от вида МкАТ, проведение сравнительного клинического исследования эффективности, включающего изучение ФК, без предварительного формального сравнительного ФК-исследования может быть затруднительным, особенно при отсутствии опыта применении у биоаналогичного (биоподобного) МкАТ у человека и потенциальной ограниченности полученных доклинических данных in vivo. Подобные исследования допускаются в индивидуальным порядке на основании полученного профиля показателей качества и доклинического профиля.
5.2. Фармакодинамика
Для определенных МкАТ и некоторых показаний свой вклад в исследования сопоставимости могут вносить ФД-параметры. В зависимости от МкАТ и доступности ФД-конечных точек теоретически возможны следующие сценарии:
5.2.1. Фармакодинамические маркеры как вспомогательное подтверждение сопоставимости.
По возможности в ФК-исследования следует включать ФД-конечные точки. Это позволяет получить ценные сведения о совокупной сопоставимости. ФД-маркеры представляют наибольшую ценность, если они позволяют обнаруживать незначительные различия и если их удается определить с высокой прецизионностью. Рекомендуется использовать несколько ФД-маркеров одновременно (при наличии). При фармакодинамическом анализе, как правило, ощущается недостаток специальных ФД-конечных точек. Поэтому основной упор необходимо сделать на доклиническую ФД-оценку, например, на испытания in vitro.
5.2.2. ФД-маркеры как опорное подтверждение сопоставимости.
Заявители должны всегда работать над поиском зависимости "доза - концентрация - эффект" или "время - эффект", поскольку такой подход при его успешности служит адекватным подтверждением сопоставимости при условии, что выбранные дозы находятся на линейной части кривой "доза - эффект".
Чтобы принять ФД-маркеры в качестве основного подтверждения клинической сопоставимости, должны быть соблюдены следующие условия:
показана четкая зависимость "доза - эффект";
по меньшей мере, один ФД-маркер является принятым суррогатным маркером и может объяснить исход пациента в такой степени, что подтверждение аналогичного эффекта с ФД-маркером будет обеспечивать аналогичное влияние на переменную клинического исхода.
Если вышеуказанное не выполняется, следует перейти к шагу 2 (к клинической эффективности).
Если в качестве основного аргумента в пользу аналогичности предполагается использовать ФД-маркеры, этот шаг рекомендуется обсудить с уполномоченными органами. Обсуждение должно включать предложенную величину границы эквивалентности и ее клиническое обоснование с точки зрения достаточности клинически значимых различий.
Сравнительное исследование с однократным или многократным введением в насыщающей части кривой "доза - концентрация - эффект", скорее всего не позволит обнаружить различия в активности (при наличии) таковой, а доза, соответствующая линейной части кривой "доза - эффект", может привести к назначению пациентам слишком низких доз. Известно, что данные о зависимости "доза-эффект" в отношении оригинального (референтного) МкАТ могут отсутствовать, а введение пациентам относительно низких доз МкАТ в худшем случае может привести к сенсибилизации и выработке антимоноклональных антител и, впоследствии, к устойчивости к терапии. Однако для некоторых оригинальных (референтных) МкАТ такие исследования в определенных клинических условиях допустимы.
5.3. Шаг 2. Клиническая эффективность
Если дозосравнительные и высокочувствительные ФД-исследования, убедительно подтверждающие клиническую сопоставимость, провести невозможно, аналогичную клиническую эффективность между биоаналогичным (биоподобным) лекарственным препаратом и оригинальным (референтным) лекарственным препаратом необходимо подтвердить в ходе достаточно мощного, рандомизированного, параллельного сравнительного клинического исследования, предпочтительно двойного слепого и исследования эквивалентности.
Для большинства клинических состояний, при которых МкАТ разрешены к применению, разработаны отдельные руководства для подтверждения их эффективности. Однако в некоторых случаях в целях подтверждения сопоставимости требуется отклониться от этих руководств (выбор конечной точки, сроки анализа конечной точки, характер или дозы сопутствующей терапии и т.п.). Такие отклонения требуют научного обоснования с позиций предложенной клинической концепции, направленной на установление биоаналогичности, с использованием ФД-маркеров, клинических исходов или и тех и других. Основополагающий принцип заключается в подтверждении аналогичной эффективности и безопасности по отношению к оригинальному (референтному) лекарственному препарату, а не пользы для пациента per se, которая ранее была установлена для оригинального (референтного) лекарственного препарата. Таким образом, в целях повышения прецизионности и облегчения интерпретации полученных результатов в целом целесообразно выбрать наиболее чувствительную популяцию пациентов и клиническую конечную точку, способные выявлять зависящие от препарата различия (при наличии таковых) и в то же время сводящие к минимуму факторы, зависящие от пациента и заболевания. Например, ответы пациентов с различной тяжестью заболевания или получавших различные виды лечения могут различаться, поэтому различия между сравниваемыми группами могут оказаться трудно интерпретируемыми, таким образом может оказаться неясным, обусловлены ли обнаруженные различия факторами, зависящими от пациента и заболевания, или же различиями между биоаналогичным (биоподобным) МкАТ и оригинальным (референтным) МкАТ.
Сопоставимость необходимо подтвердить на научно обоснованной достаточно чувствительной клинической модели и в таких же условиях исследования (независимо от регистрационного статуса), а заявитель обязан обосновать, что модель с точки зрения эффективности и безопасности является подходящей и чувствительной для подтверждения сопоставимости по заявленному показанию. Исследования сопоставимости не должны снижать безопасность для пациентов, а пациенты должны подвергаться лечению только по медицинским показаниям. Если для выявления значимых различий достаточно чувствительные клинические точки отсутствуют, заявитель обязан принять дополнительные меры, позволяющие получить достаточную чувствительность совокупного объема клинических данных, полученных в результате проведения исследования. Например, в целях достоверного установления сопоставимости исследование можно объединить с исследованием при многократном введении. Заявитель также вправе изучать фармакодинамические маркеры в дополнение к клиническим конечным точкам.
Проведение клинических исследований у особых групп пациентов (например, детей и пожилых), как правило, не требуется, поскольку конечной целью программы разработки является установление сопоставимости, вследствие чего выбор основной популяции пациентов определяется требованиями однородности и чувствительности.
В отсутствие внутренних (intrinsic) различий в исследования допускается включать пациентов из неевропейских стран, однако это может повысить гетерогенность результатов. Для предварительного установления границы эквивалентности могут потребоваться сведения об эффективности и безопасности оригинального (референтного) МкАТ в определенном регионе (местности). Если в исследование включены пациенты из различных регионов мира, в целях подтверждения непротиворечивости общему эффекту необходимо, как правило, проводить стратификацию и анализ подгрупп. Стратегии диагностики и лечения должны быть сопоставимыми в целях предотвращения влияния внешних факторов.
5.3.1. Дополнительные условия для противоопухолевых МкАТ.
Установление аналогичной клинической эффективности и безопасности биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ может оказаться особенно затруднительным для противоопухолевых препаратов. Рекомендуемой конечной точкой для подтверждения эффективности по онкологическому показанию является или выживаемость без прогрессирования и без заболевания (ВБП/ВБЗ или PFS/DFS), или общая выживаемость (ОВ или OS). Такие клинические точки необходимы в целях подтверждения пользы нового противоопухолевого препарата, однако они могут быть недостаточно подходящими или чувствительными для установления сопоставимости биоаналогичного (биоподобного) МкАТ оригинальному (референтному) препарату МкАТ, поскольку на них могут влиять различные факторы, не обусловленные различиями между биоаналогичным (биоподобным) МкАТ и оригинальным (референтным) МкАТ (например, опухолевая нагрузка, функциональный статус, предыдущая терапия, особенности клинического течения, последующая терапия (для ОВ (OS)) и т.п.) Поэтому они могут оказаться непригодными для установления аналогичной эффективности биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ.
Цель исследования сопоставимости - подтверждение аналогичной эффективности и безопасности по отношению к оригинальному (референтному) лекарственному препарату, а не пользы для пациента per se, которая ранее была установлена для оригинального (референтного) лекарственного препарата. В целях повышения прецизионности в целом целесообразно выбрать наиболее чувствительную популяцию пациентов и клиническую конечную точку, способные выявлять зависящие от препарата различия (при наличии) и в то же время сводящие к минимуму факторы, зависящие от пациента и заболевания. Допускается провести клиническое исследование в однородной популяции пациентов, выбрав в качестве первичной клиническую конечную точку, измеряющую активность. Примером может служить суммарная эффективность терапии (СЭТ или ORR, доля пациентов, у которых отмечен полный или частичный ответ (ПО и ЧО или CR и PR)). В некоторых случаях целесообразно определять СЭТ (ORR) в определенной временной точке (например, СЭТ (ORR) на n-ном месяце), долю изменения массы опухоли от исходного ее уровня или патологический полный ответ (пПО или pCR) в определенных клинических условиях. Заявители должны провести стандартизированную оценку и четкое определение конечных точек среди пациентов в соответствующие интервалы времени. По возможности необходимо регистрировать ВБП (PFS) и ОВ (OS). Ввиду множества факторов, влияющих на выживаемость, не связанных со свойствами биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ, общепризнано, что данные по выживаемости следует интерпретировать с осторожностью. Однако в тех случаях, когда использование ВБП (PFS) более чувствительно, чем СЭТ (ORR) в качестве конечной точки, его и предпочтительно использовать, несмотря на возможное удлинение исследования.
В поисковых исследованиях допускается изучать новые конечные точки (например, время до ответа), которые могут служить дополнительным подтверждением биоаналогичности.
5.4. Клиническая безопасность
На протяжении всей клинической разработки важную роль играет клиническая безопасность, она подлежит изучению как в ходе начальных ФК-оценок и (или) ФД-оценок, так и как часть ключевых клинических исследований, направленных на установление сопоставимости (подобия). Вид, тяжесть и частоту нежелательных реакций между биоаналогичным (биоподобным) МкАТ и оригинальным (референтным) МкАТ, особенно описанных для последнего, необходимо подвергать тщательному сравнению. Если единые подходы к определению параметров безопасности не выработаны (например, оценка кардиотоксичности), рекомендуется использовать те же подходы, которые применялись в программе разработки оригинального (референтного) МкАТ (при наличии сведений), или подходы, применяемые в процессе пострегистрационного наблюдения. В качестве дополнительного подтверждения клинической сопоставимости допускается также осуществлять сравнение фармакологически опосредованных нежелательных реакций (например, кардиотоксичности), то есть фармакодинамических маркеров, затрагивающих безопасность. Их анализ проводят в соответствии с принципами, описанными в отношении ФД-маркеров эффективности.
Если для получения основного подтверждения эквивалентности в части клинической эффективности достаточно провести сравнительные высокочувствительные ФД-исследования, заявители должны представить достаточные аргументы в пользу клинической безопасности, включая иммуногенность. Данные по безопасности в сравнении с активным контролем, как правило, получают на предрегистрационном этапе. На них оказывают влияние свойства МкАТ и количество пациентов, которым вводили препарат, а также продолжительность терапии. Длительность наблюдения за безопасностью на предрегистрационном этапе необходимо обосновать.
Часть данных по безопасности, а также дополнительные данные по безопасности может потребоваться набрать на пострегистрационном этапе. Вероятность выявления редких явлений (например, прогрессирующей многоочаговой лейкоэнцефалопатии) на предрегистрационном этапе незначительна. В связи с этим в момент регистрации заявитель должен описать свою деятельность по фармаконадзору и управлению рисками на пострегистрационном этапе. Вместо прямого сопоставления с оригинальным (референтным) лекарственным препаратом, как правило, требуется схожая с последним деятельность по фармаконадзору, поскольку сравнительные данные будут трудно интерпретируемыми в силу редкости их возникновения, что приведет к неточности оценки выявленных различий.
Заявители должны отразить, каким образом будет осуществляться повторное лечение пациентов. На момент подачи заявления о регистрации лекарственного препарата необходимо представить концепцию о способе систематического определения безопасности многократного введения лекарственного препарата пациентам (например, по онкологическим показаниям, когда пациенты подвергаются нескольким циклам терапии). В соответствующих случаях рекомендуется продлить клиническое исследование до пострегистрационного наблюдательного исследования, чтобы охватить весь цикл лечения.
Ввиду таких клинических последствий, как снижение эффективности, а также вероятной резистентности к дальнейшему лечению оригинальным (референтным) лекарственным препаратом необходимо проводить систематическое сравнительные изучение иммуногенности. Целесообразно (по возможности) не включать пациентов, которым ранее вводили оригинальное (референтное) МкАТ, или заранее предусмотреть анализ подгруппы пациентов, ранее получавших лечение (с целью установления влияния предшествующего лечения на развитие иммуногенности), поскольку предшествующая терапия могла послужить причиной образования антител к препарату, которые могут осложнить интерпретацию данных по безопасности и, таким образом, также снизить чувствительность выявления различий. Сравнительную оценку нежелательного иммунного ответа к биоаналогичному (биоподобному) МкАТ и оригинальному (референтному) МкАТ, как правило, осуществляют как часть клинического исследования, направленного на установление аналогичной клинической эффективности и безопасности, используя те же валидированные методики (см. главы 11 и 12 настоящих Правил). Допускается использовать подход популяционной ФК, предусматривающий редкий отбор образцов и определение концентрации препарата, а также антител к нему. Однако антитела к некоторым МкАТ лучше обнаруживаются у здоровых добровольцев, у которых в течение нескольких дней после однократного введения развивается сильный иммунный ответ. Введенная доза также является важным фактором, требующим учета при изучении иммуногенности: некоторые МкАТ при введении их в больших дозах препятствуют образованию антител, в связи с этим исследования, проведенные с низкими дозами, при их медицинской оправданности, более чувствительны для сравнения иммунного ответа к биоаналогичному (биоподобному) лекарственному препарату и оригинальному (референтному) лекарственному препарату.
Изучение нежелательной иммуногенности особенно необходимо, если в производстве биоаналогичного (биоподобного) МкАТ использовалась иная по сравнению с оригинальным (референтным) МкАТ экспрессирующая конструкция, что может, к примеру, приводить к образованию значимых показателей качества, которые не обнаруживались у оригинального (референтного) лекарственного препарата (например, новая посттрансляционная структурная модификация), способных привести к повышенной иммуногенности. Это особенно необходимо, если опыт использования такой конструкции у человека ограничен. Такие подходы рекомендуется заранее согласовывать с уполномоченными органами.
Повышенная по отношению к оригинальному (референтному) МкАТ иммуногенность может стать основным препятствием при анализе отношения пользы и риска, она также ставит под сомнение биоаналогичность. Однако возможна и меньшая иммуногенность биоаналогичного (биоподобного) МкАТ, которая не препятствует биоаналогичности. В таком случае при анализе эффективности всей популяции пациентов можно прийти к заключению, что биоаналог (биоподобный препарат) более эффективен (поскольку у меньшего количества пациентов развился иммунный ответ, в связи с чем у большего числа из них может проявиться терапевтический эффект биоаналогичного (биоподобного) МкАТ). В связи с этим рекомендуется предусмотреть дополнительный поисковый анализ эффективности и безопасности в подгруппе пациентов, у которых в ходе клинического исследования антитела к препарату не образовались. Анализ такой подгруппы будет способствовать установлению того, что эффективность биоаналогичного (биоподобного) МкАТ и оригинального (референтного) МкАТ без учета иммунного ответа в целом аналогична.
На пострегистрационном этапе могут потребоваться дополнительные данные по долгосрочной иммуногенности и безопасности, например, в случаях небольшой продолжительности исследования, направленного на установление аналогичной клинической эффективности. В плане управления рисками следует проанализировать необходимость получения дополнительных данных по долгосрочной иммуногенности и безопасности. Если такие данные требуются, заявители не должны ограничиваться стандартными мерами по фармаконадзору, а провести пострегистрационное исследование безопасности. Относительно безопасности и иммуногенности по различным показаниям, разрешенным для оригинального (референтного) МкАТ и заявленным для биоаналогичного (биоподобного) МкАТ, в целях получения более подробных данных по безопасности в зависимости от показаний может потребоваться реализовать пострегистрационную концепцию, описанную в разделе 5.
6. Экстраполяция показаний
Экстраполяция данных клинической эффективности и безопасности на другие показания оригинального (референтного) МкАТ, отдельно не изучавшихся в ходе клинической разработки биоаналогичного (биоподобного) МкАТ, на основании совокупного подтверждения сопоставимости (подобия), полученного по результатам сравнительных исследований, и при достаточном обосновании возможна. Если основное подтверждение сопоставимости основывается на ФД, а заявленные показания реализуются за счет различных механизмов действия (либо имеется неопределенность в данном вопросе), заявители должны представить соответствующие данные, обосновывающие экстраполяцию на все заявленные показания к применению. Заявители должны обосновать такую экстраполяцию всесторонним анализом имеющейся литературы, включая вовлеченные рецепторы антигенов и механизмы действия.
Например, если оригинальное (референтное) МкАТ применяется как в качестве иммуномодулирующего, так и противоопухолевого антитела научное обоснование экстраполяции между двумя (или более) показаниями более затруднительно. Основу такой экстраполяции образует большая база данных по качеству и доклиническим исследованиям, включая методы количественного определения активности и in vitro методы, устанавливающие функциональность молекулы, а также соответствующие клинические данные, описанные ниже. Возможность экстраполяции данных по безопасности, включая иммуногенность, также требует тщательного рассмотрения и может потребовать вовлечения более специфичных исследований (см. разделы 5 и 7). Относительно механизма действия: снижение числа иммунных клеток, к примеру, при различных состояниях может реализовываться за счет различных механизмов. Например, АЗКЦ более значима при одних показаниях, чем при других. Для представления более глубоких данных о механизме действия целесообразно провести литературный поиск с целью установления известных фактов, например, о потенциальном ингибировании передачи сигнала оригинальным (референтным) лекарственным препаратом, которое не изучалось в испытаниях АЗКЦ (КЗЦ), в особенности прямой индукции апоптоза. Это может расширить знания о потенциальных аргументах, которые могут быть использованы для подтверждения сопоставимости на молекулярном уровне.
7. Фармаконадзор
В соответствии с актами, входящими в право Союза, в ходе процедуры регистрации заявитель должен представить план управления рисками и план фармаконадзора. Допускается осуществлять деятельность по минимизации рисков, разработанную для оригинального (референтного) лекарственного препарата, а также включить ее в план управления рисками биоаналогичного (биоподобного) лекарственного препарата.
В целях углубленного анализа вопросов безопасности, описанных выше, на момент подачи заявления на регистрацию заявители должны представить всеобъемлющую концепцию дальнейшего изучения безопасности на пострегистрационном этапе, включая следующие аспекты:
в отсутствие обоснований - безопасность по показаниям, разрешенным для оригинального (референтного) МкАТ, зарегистрированным на основании экстраполяции данных по эффективности и безопасности, включая данные о долгосрочной безопасности;
возникновение редких и особо серьезных нежелательных явлений, описанных и прогнозируемых на основании фармакологических свойств оригинального (референтного) МкАТ. План фармаконадзора должен соответствовать выявленным и потенциальным рискам и содержаться в спецификации по безопасности МкАТ сравнения в дополнение к соответствующим данным о биоаналогичных (биоподобных) лекарственных препаратах соответственно;
выявление новых сигналов по безопасности, подобно прочим биологическим лекарственным препаратам;
деятельность по получению дополнительных данных по иммуногенности (при необходимости).
Такая концепция может потребовать превысить стандартные действия по фармаконадзору и потребовать более активной деятельности по фармаконадзору, например, ведения реестров или базы данных больших групп пациентов, в которых ввод данных осуществляется стандартизованно, обеспечивая правильность и постоянство регистрации (анализа). Кроме того, рекомендуется участие в уже имеющихся реестрах, что должно составлять часть плана управления рисками. Достаточность таких предложений должна оцениваться в свете данных по безопасности на момент регистрации, совокупности данных по результатам исследований сопоставимости и профиля безопасности оригинального (референтного) МкАТ. Необходимо четко оценить необходимость в дополнительной деятельности по минимизации рисков с учетом требований к оригинальному (референтному) лекарственному препарату.
Необходимо особо тщательно идентифицировать подозреваемые нежелательные реакции на биологические лекарственные препараты с точки зрения их производства. В связи с этим следует принять все необходимые меры для четкой идентификации биологического лекарственного препарата, являющегося объектом отчета о нежелательных реакциях, с точным указанием его наименования и номера серии.
В зависимости от особенностей обращения биоаналогичных (биоподобных) и оригинальных (референтных) лекарственных препаратов в клинической практике на национальном уровне возможен переход с препарата на препарат или взаимная замена препаратов, содержащих определенное МкАТ. В связи с этим заявителям рекомендуется учитывать эти обстоятельства в плане управления рисками.
Глава 15.4. Доклинические и клинические исследования
биоаналогичного (биоподобного) лекарственного препарата
рекомбинантного эритропоэтина
1. Вступление
В настоящей главе изложены требования к доклиническим и клиническим исследованиям лекарственных препаратов, содержащих рекомбинантный человеческий эритропоэтин, заявленный при регистрации в качестве биоаналогичного (биоподобного) уже присутствующим на рынке лекарственным препаратам. В разделе, посвященном доклиническим исследованиям, представлены рекомендации для оценки фармакотоксикологических свойств. В разделе, посвященном клиническим исследованиям, представлены требования для оценки фармакокинетических и фармакодинамических свойств, безопасности и эффективности и составления плана управления рисками. Кроме того, представлены критерии, необходимые для экстраполяции клинических данных на другие показания, утвержденные для оригинального (референтного) препарата.
Эритропоэтин человека является гликопротеином, состоящим из 165 аминокислот, который секретируется почками и стимулирует продукцию эритроцитов. Препараты, которые являются аналогами эндогенного эритропоэтина и которые производятся на основе технологии рекомбинантной ДНК с использованием клеток млекопитающих в качестве системы экспрессии, получили название эпоэтины. Применяемые в клинической практике эпоэтины имеют последовательность аминокислот, которая соответствует эндогенному эритропоэтину, однако могут различаться между собой по степени гликозилирования. От качественных и количественных характеристик гликозилирования эпоэтинов зависят фармакокинетические свойства препарата, а также его эффективность и безопасность, в том числе и иммуногенность. Для характеристики белка используют физико-химические и биологические методы.
Препараты, содержащие эпоэтины, применяются для лечения симптоматических анемий у больных с хронической почечной недостаточностью, анемий, которые развиваются под влиянием химиотерапии у больных онкологическими заболеваниями, для увеличения сбора аутологичной крови, у пациентов, участвующих в программе предварительного донорства. Механизм действия эпоэтинов одинаков при всех одобренных показаниях, однако дозы, необходимые для достижения терапевтического эффекта, могут значительно различаться (наиболее высокие дозы применяются при онкологических заболеваниях). Препараты на основе эпоэтинов вводятся внутривенно или подкожно.
Препараты рекомбинантных эпоэтинов имеют широкий терапевтический диапазон и, как правило, хорошо переносятся при условии надлежащего контроля стимуляции эритропоэза по показателю уровня гемоглобина и скорости его повышения. Скорость повышения уровня гемоглобина варьирует и зависит не только от дозы и схемы лечения, но и от таких факторов, как содержание железа, исходный уровень гемоглобина и уровень эндогенного эритропоэтина, а также наличия сопутствующих заболеваний (например, воспалительных).
Чрезмерный фармакодинамический ответ может приводить к развитию артериальной гипертензии и тромбоэмболических осложнений. Кроме того, при подкожном введении эпоэтина у больных с почечной анемией зарегистрированы случаи развития истинной парциальной красноклеточной аплазии, вызванной выработкой нейтрализующих антител к эпоэтину. Учитывая, что развитие истинной парциальной красноклеточной аплазии встречается редко и для ее развития требуется длительное, в течение нескольких месяцев или лет применение препарата, вероятность ее выявления в ходе предрегистрационных исследований крайне низка. Кроме того, у некоторых групп пациентов особую важность приобретает ангиогенное и онкогенное действие эритропоэтина.
Регистрационное досье на новый лекарственный препарат, содержащий эритропоэтин, заявленный в качестве подобного ранее зарегистрированному на территории государств-членов оригинальному (референтному) препарату, должно содержать данные, демонстрирующие сопоставимое качество, безопасность и эффективность заявленного к регистрации препарата оригинальному (референтному) лекарственному препарату, зарегистрированному в Союзе.
2. Сфера применения
Настоящая препаратспецифичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих эритропоэтин.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Основные указания
4.1. Доклинические исследования
Доклинические исследования проводятся до начала клинических исследований. Основной целью сравнительных доклинических исследований является выявление возможных различий фармако-токсикологических свойств между биоаналогичным (биоподобным) и оригинальным (референтным) препаратом, а не простое доклиническое изучение эффекта препарата как такового. При этом выбор подхода (программа проведения и объем исследований) должен быть полностью обоснован в доклиническом обзоре (модуль 2.4 регистрационного досье).
Изучение фармакодинамики
Исследования in vitro
Для выявления возможных различий в ответной реакции на биоаналогичный (биоподобный) и оригинальный (референтный) препараты должен быть проведен ряд сравнительных биологических исследований in vitro (например, изучение способности специфически взаимодействовать с соответствующим рецептором, оценка способности стимулировать пролиферацию соответствующих линий клеток). Многие из этих данных могут быть доступны в процессе сравнительного изучения показателей качества сравниваемых препаратов.
Исследования in vivo
Сравнительное изучение эритрогенной активности биоаналогичного (биоподобного) и оригинального (референтного) препаратов проводится в соответствующих исследованиях на экспериментальных животных. Данные о способности препаратов стимулировать эритропоэз могут быть получены в процессе изучения хронической токсичности при многократном введении препарата или в специальном исследовании (например, при изучении специфической активности по степени стимуляции ретикулоцитоза на нормоцитемических или полицитемических мышах, в соответствии с Фармакопеей Союза, подобные данные могут быть получены на этапе изучения качества препарата).
Изучение токсичности
Изучение токсичности должно включать как минимум одно сравнительное исследование хронической токсичности (многократное введение одной дозы препарата) на релевантном виде животного (например, крысах). Продолжительность исследований должна быть не менее 4 недель. Исследования токсичности проводятся с учетом требований, указанных в главах 5.3 и 15.2 настоящих Правил, посвященных вопросам изучения безопасности терапевтических биотехнологических препаратов. В рамках исследования токсичности биоаналогичного (биоподобного) препарата при многократном введении препарата должна быть выполнена соответствующая оценка токсикокинетики, а также должна быть изучена и определена возможность выработки к нему антител (глава 11 настоящих Правил).
Кроме того, должны быть представлены данные о местной переносимости препарата, полученные по меньшей мере на одном виде животных. При этом рекомендуется определить местную переносимость в рамках описанного выше исследования токсичности при многократном введении препарата. Фармакологические исследования безопасности, исследования токсического действия на репродуктивную систему, исследования мутагенности и канцерогенности не входят в состав обязательных стандартных требований к доклиническим исследованиям подобных биологических лекарственных препаратов, содержащих в качестве фармацевтической субстанции эритропоэтин.
4.2. Клинические исследования
Изучение фармакокинетики
Изучение фармакокинетических свойств проводится в сравнительном перекрестном исследовании биоаналогичного (биоподобного) и оригинального (референтного) препаратов при однократном применении с использованием разных путей введения (как правило, подкожного и внутривенного). Перекрестные исследования целесообразно проводить с участием здоровых добровольцев. Для изучения фармакокинетики и проведения сравнительного анализа необходимо подобрать дозы, которые будут соответствовать наиболее крутой части кривой зависимости "доза - эффект". Изучение фармакокинетических свойств препаратов включает параметры AUC, Cmax и T1/2 или C1/F. Пределы эквивалентности должны быть определены заранее и должным образом обоснованы. При разработке программы исследований необходимо учитывать различия в показателях T1/2 при внутривенном и подкожном введении и зависимости клиренса от дозы препарата.
Изучение фармакодинамики
Исследование фармакодинамики предпочтительно проводить в процессе сравнительного изучения фармакокинетики. Необходимо выбрать дозу, которая должна находиться в области линейной восходящей части кривой зависимости "доза - эффект". В качестве фармакодинамического маркера при оценке активности эпоэтина в исследованиях с однократным введением препарата наибольшую значимость имеет определение количества ретикулоцитов. При этом количество ретикулоцитов не является суррогатным маркером при оценке эффективности эпоэтина и поэтому не может быть использовано в качестве конечной точки при клиническом изучении эффективности препарата.
Изучение эффективности
Сходство (подобие) клинической эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов должно быть продемонстрировано в сравнительных рандомизированных клинических исследованиях достаточной статистической мощности, проведенных на параллельных группах пациентов. Учитывая, что дозы и фармакокинетические показатели при внутривенном и подкожном введении препарата различаются, необходимо провести изучение эффективности и подтвердить сопоставимость препаратов при обоих путях введения. Это может быть выполнено при проведении отдельных клинических исследований для каждого способа введения или путем проведения одного клинического исследования для одного способа введения с адекватной экстраполяцией соответствующих данных для другого способа введения.
Подтверждающие исследования для исключения систематической ошибки следует проводить в виде двойного слепого клинического исследования. Если проведение данных исследований невозможно, то при распределении пациентов по группам сравнения необходимо предусмотреть маскирование данных о схемах лечения от медицинского персонала, вовлеченного в процесс принятия решений (например, при подборе дозы лекарственного препарата).
Для оценки эффективности эпоэтинов необходимо выбрать наиболее чувствительную популяцию пациентов (в частности, таковой является популяция больных с дефицитом эндогенного эритропоэтина, по сравнению с больными без дефицита). Кроме того, чувствительность пациентов к терапевтическому действию эпоэтина зависит от чувствительности костного мозга к эритропоэтину. Больные с симптоматической анемией, вызванной почечной недостаточностью при условии отсутствия у них серьезных осложнений (например, тяжелых хронических заболеваний, кровотечений или интоксикации солями алюминия), которые могут повлиять на эффективность препарата, являются наиболее оптимальной популяцией для изучения эффективности эритропоэтинов поскольку данная категория пациентов является наиболее трудно отвечающей на терапию. При этом необходимо исключить развитие анемии в результате других причин. Дозы эпоэтинов, которые вводятся для достижения нормального уровня гемоглобина и его поддержания, различаются при назначении больным, находящимся на диализе, и преддиализным больным. Поэтому больных из указанных групп не рекомендуется смешивать в одном клиническом исследовании.
Для демонстрации подобия препаратов могут быть использованы представленные ниже варианты дизайна проведения исследований. Заявитель вправе выбрать любой из предложенных вариантов дизайна исследований или модифицировать его, при условии представления значимого научного обоснования выбранного подхода.
Демонстрация эффективности двух путей введения препарата
Эффективность обоих (подкожного и внутривенного) путей введения препарата может быть продемонстрирована при проведении двух отдельных исследований. Наиболее информативным с точки зрения получения данных о биоаналогичных (биоподобных) эритропоэтинах является комбинированное исследование, которое включает изучение подкожного пути введения препарата в "фазе коррекции" (например, на группе преддиализных больных) и изучение при внутривенном введении в "поддерживающей фазе" (например, на группе больных, находящихся на диализе).
В "фазе коррекции" проводится оценка динамики ответа и определяется схема дозирования препарата при коррекции анемии, также в данной фазе оценивается профиль безопасности, связанный с фармакодинамикой препарата. Исследование проводится среди больных, которые ранее никогда не получали препараты эпоэтинов или не получали данные препараты в течение достаточно длительного срока (как минимум около 3 месяцев) и которым не проводилось переливание крови. В тех случаях, когда больные получали препараты эпоэтинов пролонгированного действия (например, пэгилированный эпоэтин), период после их последнего применения должен быть еще более продолжительным.
Исследование в "поддерживающей фазе" является более чувствительным для выявления различий в уровне проявления биологической активности биоаналогичного (биоподобного) и оригинального (референтного) препаратов, хотя исследования в "фазе коррекции" также являются достаточно информативными (дискриминативными). При разработке стратегии исследования эффективности применения препаратов в "поддерживающей фазе" необходимо свести к минимуму исходную гетерогенность исследуемой группы пациентов, а также обеспечить учет возможного проявления терапевтического эффекта предшествующего лечения. Для пациентов, которых планируется включить в исследование по использованию препаратов в "поддерживающей фазе", должна быть правильно определена доза, установленная относительно препарата сравнения (доза эритропоэтина, обеспечивающая стабильный уровень гемоглобина в целевом диапазоне на фоне стабильной дозы эритропоэтина и стабильного режима введения при отсутствии гемотрансфузий) в течение длительного времени (обычно не менее 3 месяцев). После этого все включенные в исследование пациенты должны быть рандомизированы по группам, которые будут получать биоаналогичный (биоподобный) или оригинальный (референтный) препарат, сохраняя установленные до рандомизации дозу, схему и способ применения эритропоэтина.
Как вариант, при представлении объективного полного обоснования возможно проведение раздельных исследований эффективности внутривенного и подкожного введения препаратов в "поддерживающей фазе".
В ходе проведения обоих исследований необходимо тщательно подобрать дозы эпоэтина, необходимые для достижения в "фазе коррекции" или поддержания в "поддерживающей фазе" целевого уровня концентрации гемоглобина. Алгоритм подбора дозы эпоэтина должен соответствовать правилам надлежащей клинической практики Союза, утверждаемым Комиссией, и проводиться одинаково в обеих группах.
Предпочтительными первичными конечными точками исследований в "фазе коррекции" является "доля пациентов с положительным ответом на лечение" (доля пациентов в процентах, у которых количество гемоглобина повысилось до установленного уровня) или "изменение уровня гемоглобина". При проведении исследований в "поддерживающую фазу" первичными конечными точками являются "доля пациентов с сохранением ответа на лечение" (доля (в процентах) пациентов, у которых зафиксировано поддержание количества гемоглобина на заранее установленном целевом уровне) или "изменение уровня гемоглобина". Следует учитывать, что подбор дозы эпоэтина, которая необходима для повышения гемоглобина до установленного уровня, снижает чувствительность первичных конечных точек, которые определяются по динамике содержания гемоглобина, для выявления различий по показателям эффективности между сравниваемыми группами. Поэтому доза эпоэтина должна быть включена в состав комбинированной первичной конечной точки в обоих типах исследований.
Результаты оценки первичных конечных точек при изучении эффективности препарата должны быть собраны в течение определенного периода исследования. Четырехнедельный период с 5-го по 6-й месяцы исследований как "фазы коррекции", так и "поддерживающей фазы" является оптимальным как для предотвращения возникновения эффектов переноса, связанных с лечением на исходном уровне, так и для полной оценки возможных различий между обеими конечными точками при наличии стабильных уровней гемоглобина и одинаковых дозировок эритропоэтина. Если оценка первичной конечной точки проводится в более короткий промежуток времени, необходимо представить обоснование того, что данный период исследования позволит выявить потенциальные различия эффективности сравниваемых препаратов.
Пределы эквивалентности для обеих сопутствующих конечных точек должны быть определены и должным образом обоснованы заранее, так как они являются основой, обеспечивающей исследованиям статистическую значимость. Если в качестве первичной конечной точки используется изменение уровня гемоглобина в сравнении с исходным, то рекомендуемый порог эквивалентности составляет +/- 0,5 г/дл. Необходимость в переливании крови следует включить в качестве важной вторичной конечной точки.
Второй подход для доказательства сходства эффективности обоих путей введения препарата включает сравнительную оценку изучения эффективности при одном пути введения препарата с помощью сравнительного клинического исследования на пациентах и представления сопоставимых фармакокинетических (фармакодинамических) данных связующего исследования при однократном и многократном введении препарата пациентам из чувствительной к эритропоэтину группы (например, здоровым добровольцам), а затем экстраполяции этих данных на второй путь введения. При многократном введении препарата оценка фармакокинетики (фармакодинамики) проводится с использованием фиксированной дозы эпоэтина в пределах широты терапевтического диапазона. В качестве первичной конечной точки фармакодинамики необходимо использовать показатели изменения уровня гемоглобина, а длительность исследования должна составлять не менее 4 недель.
Для подкожного пути введения обязательной является сравнительная оценка иммуногенности и наиболее предпочтительными при данном подходе являются проведение клинического исследования при подкожном пути введения препарата и представление данных связующего исследования фармакокинетики (фармакодинамики) для внутривенного введения препарата.
В случае когда пациенты включаются в группы, получающие препараты подкожно на протяжении 12 месяцев, они могут быть использованы для сравнительного изучения иммуногенности в течение 12 месяцев. После этого, пациентам, которые получали оригинальный (референтный) препарат, назначают лечение биоаналогичным (биоподобным) препаратом, наблюдение проводят еще в течение 6 месяцев, что позволяет расширить базу данных по оценке безопасности и иммуногенности исследуемого биоаналогичного (биоподобного) препарата. Если нет возможности проведения исследований в соответствии с данным дизайном, при подготовке программы исследования выбор популяции и конечных точек исследования необходимо проводить с учетом указанных рекомендаций.
Изучение эффективности одного пути введения препарата
Если предполагается, что препарат будет регистрироваться только для одного пути введения, необходимо проведение исследования фармакокинетики (фармакодинамики) при однократном введении препарата, используемого в одной из фаз ("фазы коррекции" или "поддерживающей фазы"), выбранной для данного пути введения. При разработке программы исследования, выборе популяции и первичных конечных точек исследования необходимо руководствоваться соответствующими рекомендациями настоящих Правил.
Отсутствие данных для одного из путей введения должно быть четко указано в общей характеристике лекарственного препарата для данного биоаналогичного (биоподобного) эритропоэтина.
4.3. Исследование безопасности
Обычно данных о безопасности, полученных при изучении эффективности на предрегистрационном этапе исследования, достаточно для создания адекватной базы данных предмаркетинговой безопасности. Основными нежелательными явлениями, представляющими особый интерес, при применении эпоэтинов являются развитие (усугубление) артериальной гипертензии и тромбоэмболические нарушения.
Исследование иммуногенности проводится в соответствии с главой 11 настоящих Правил, согласно которой для регистрации препарата необходимо представить результаты, полученные в исследованиях по оценке иммуногенности препарата в течение не менее 12 месяцев. При отсутствии стандартизованных методов оценки иммуногенности для правильной интерпретации результатов требуются сравнительные данные изучения иммуногенности оригинального (референтного) препарата. Период сравнительного изучения иммуногенности должен включать как минимум 12 месяцев. При изучении иммуногенности в более короткие сроки необходимо представить убедительные аргументы в пользу того, что это не повлияет на объективную оценку иммуногенного потенциала биоаналогичного (биоподобного) эпоэтина.
При изучении иммуногенности необходимо использовать современные валидированные и высокочувствительные методы определения антител, которые формируются в ранние сроки после введения препарата (антител, характеризующихся низкой аффинностью, главным образом IgM), и антител, определяемых в поздние сроки (обладающих высокой аффинностью). Кроме того, необходимо провести дополнительную характеристику выявленных антител и в первую очередь оценить их нейтрализующую активность. При этом рекомендуется сохранять образцы, которые получены при применении препаратов в "фазе коррекции" и "поддерживающей фазе". Учитывая, что нейтрализующие антитела и истинная парциальная красноклеточная аплазия при лечении эпоэтином встречаются редко, вероятность их выявления на этапе клинических исследований в предрегистрационном периоде низкая. Однако если они все-таки будут выявлены при клинических исследованиях, это будет свидетельствовать о серьезных проблемах, связанных с безопасностью применения препарата. Несмотря на то, что значение связывающих ненейтрализующих антител до конца не ясно, тем не менее определение их повышенного количества или частоты выявления в ответ на введение изучаемого препарата не позволяет сделать положительный вывод о безопасности препарата и рассматривать его как биоаналогичный (биоподобный).
Иммуногенность препаратов эпоэтина выше при подкожном пути введения, чем при внутривенном. Больные с почечной недостаточностью имеют наибольший риск выработки антител к эпоэтину, которые могут инициировать развитие истинной парциальной красноклеточной аплазии. Поэтому оценка иммуногенности должна включать достаточное количество пациентов с данной патологией и подкожным введением препарата, если только подкожное введение для данной популяции пациентов не противопоказано.
4.4. План фармаконадзора
При регистрации препарата в модуль 1 регистрационного досье необходимо включить план управления рисками (план по фармаконадзору) в соответствии с правилами регистрации и экспертизы лекарственных средств для медицинского применения и правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией. В плане управления рисками (плане фармаконадзора) должны быть отражены сведения по контролю за развитием редких таких серьезных побочных реакций, как реакции, опосредованные иммуногенностью, истинная парциальная красноклеточная аплазия и оценка канцерогенного потенциала препарата.
4.5. Экстраполяция результатов исследований
на другие показания
Учитывая, что механизм действия эпоэтина является одинаковым для всех известных показаний к применению и имеется только один известный рецептор для эпоэтина, результаты, полученные при проведении исследований для доказательства эффективности и безопасности изучаемого препарата при анемии почечного происхождения, могут быть экстраполированы на другие показания к применению, указанные в общей характеристике лекарственного препарата для оригинального (референтного) препарата, использованного в качестве препарата сравнения, при том же пути введения.
Глава 15.5. Доклинические и клинические исследования
биоаналогичного (биоподобного) лекарственного препарата
рекомбинантного гранулоцитарного
колониестимулирующего фактора
1. Введение
В настоящей главе изложены указания по демонстрации сопоставимости двух лекарственных препаратов, содержащих в качестве действующего вещества рекомбинантный гранулоцитарный колониестимулирующий фактор (рГ-КСФ).
В разделе о доклинических исследованиях обосновывается необходимость соответствующих фармакодинамических и токсикологических исследований. В разделе о клинических исследованиях приведены рекомендации о подходящих видах исследований фармакодинамики и фармакокинетики, определении параметров эффективности и безопасности, необходимых для демонстрации подобия двух лекарственных препаратов на основе рГ-КСФ, а также сведения о специальных мероприятиях, направленных на управление рисками. Также обсуждаются критерии экстраполяции клинических данных на другие показания, одобренные для оригинального (референтного) лекарственного препарата.
В регистрационное досье на вновь разработанный лекарственный препарат, содержащий в качестве действующего вещества рГ-КСФ, и представляемый для регистрации как биоаналогичный (биоподобный) оригинальному (референтному) лекарственному препарату, зарегистрированному в государствах-членах, должны быть включены материалы, обосновывающие доказательства сходства (подобия) указанных препаратов.
Эндогенный Г-КСФ человека является кислым гликопротеином, состоящим из одной полипептидной цепи 174 аминокислот, его молекулярная масса составляет 19,6 кДа. Молекула Г-КСФ содержит свободную сульфгидрильную группу цистеина (в положении Cys17) и две внутримолекулярные дисульфидные связи в положениях Cys36-Cys42 и Cys64-Cys74. Эндогенный Г-КСФ человека не содержит сайтов N-гликозилирования и единственная O-гликозильная группа присоединена к остатку треонина (Thr133). Углеводный остаток на Thr133 составляет около 4 процентов от общей молекулярной массы гликопротеина.
В клинической практике используются препараты, полученные с помощью технологии рекомбинантной ДНК в системе клеток Escherichia coli (филграстим) или в системе клеток яичников китайских хомячков (CHO) (ленограстим). Молекула рекомбинантного Г-КСФ, полученного в системе клеток E. coli, отличается отсутствием гликозилирования и наличием дополнительного метионина на N-конце молекулы от нативного Г-КСФ и рекомбинантного Г-КСФ, полученного в системе клеток млекопитающих. Молекула рчГ-КСФ содержит один свободный остаток цистеина и две дисульфидные связи.
Специфическое действие Г-КСФ человека реализуется в результате связывания со специфическим трансмембранным рецептором, экспрессированным на различных гемопоэтических клетках, включая стволовые клетки, предшественники мультипотентных клеток, предшественники миелоидных клеток, нейтрофилов и моноцитов. Выделяют семь мембранно-ассоциированных и одну растворимую изоформу рецептора Г-КСФ, внеклеточные лиганд-связывающие домены всех изоформ идентичны. Эффекты Г-КСФ (рекомбинантного и эндогенного) опосредованы взаимодействием с рецептором одного класса аффинности.
Антитела к уже присутствующему на рынке рчГ-КСФ, синтезированному в культуре клеток E. coli, вырабатываются не часто, поэтому не оказывают существенного влияния на безопасность и эффективность применения препаратов. Лекарственные препараты выпускаются для внутривенного и подкожного введения. Возможные факторы риска развития иммунного ответа, связанные с особенностями пациентов, не известны.
Настоящая глава подлежит применению совместно с другими главами настоящих Правил и иными актами, входящими в право Союза.
2. Сфера применения
Настоящая препаратспецифичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих рчГ-КСФ.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Основной текст указаний
4.1. Доклинические исследования
Перед началом клинических исследований должны быть проведены доклинические исследования. Такие исследования носят сравнительный характер и направлены на выявление различий между фармако-токсикологическими эффектами применения подобного лекарственного препарата и оригинального (референтного) лекарственного препарата, а не на изучение результатов лечения как таковых. Выбор способа исследования должен быть полностью обоснован в доклиническом обзоре.
Фармакодинамические исследования
Исследования in vitro
На рецепторном уровне доказательство подобия исследуемого и оригинального (референтного) препарата должно быть продемонстрировано при помощи in vitro клеточного биотеста или анализа связывания с рецептором. Также эти данные могут быть получены по результатам биотестов, проведенных для измерения активности при оценке биологических характеристик в модуле 3 регистрационного досье. Важно, чтобы методики, используемые для оценки доказательства подобия, обладали чувствительностью, достаточной для выявления различий, а также чтобы в ходе экспериментов использовалось количество разведений, достаточное для составления кривой зависимости ответа от концентрации.
Исследования in vivo
Сравнение фармакодинамических эффектов исследуемого препарата и оригинального (референтного) лекарственного препарата необходимо проводить в опытах на грызунах in vivo как с нейтропенией, так и без нее.
Исследование токсичности
Необходимо представить данные, полученные в ходе по меньшей мере одного исследования токсичности с повторным введением препарата подходящим видам животных (при этом следует учитывать этапный подход, изложенный в главе 15.2 настоящих Правил). Продолжительность исследования должна составлять не менее 28 дней. Исследование по изучению токсичности при многократном введении препарата должно включать определение фармакодинамических показателей и определение токсикокинетических показателей. Особое внимание следует уделить изучению иммунного ответа на препарат. Также необходимо представить данные о местной переносимости, полученные по меньшей мере на одном виде животных. По возможности в рамках исследования токсичности при многократном введении препарата необходимо провести исследование местной переносимости. Исследования фармакологической безопасности, репродуктивной токсичности, мутагенности и канцерогенности не являются рутинными требованиями к доклиническим исследованиям биоаналогичных (биоподобных) лекарственных препаратов, содержащих в качестве действующего вещества Г-КСФ.
4.2. Клинические исследования
Исследование фармакокинетики
Демонстрация подобия (сходства) на клиническом этапе проводится поэтапно и начинается со сравнительной оценки фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов. Изучение проводится у здоровых добровольцев в перекрестном исследовании при однократном подкожном и внутривенном введении сравниваемых препаратов. При сравнительном изучении фармакокинетических свойств в качестве основного показателя необходимо использовать AUC, в качестве дополнительных показателей Cmax и T1/2. Для оценки результатов исследований могут быть использованы общие принципы оценки биоэквивалентности.
Исследование фармакодинамики
Основным маркером фармакодинамики препаратов Г-КСФ является абсолютное количество нейтрофилов. Доза препарата при изучении фармакодинамики должна быть подобрана таким образом, чтобы она соответствовала крутой части кривой "доза - эффект", для этого может потребоваться изучение нескольких доз препаратов. Вторичной конечной точкой сравнительной оценки фармакодинамики является динамика количества CD34 клеток. Диапазон допустимого предела отклонения оцениваемых показателей должен быть обоснован с учетом данных изучения фармакодинамики оригинального (референтного) препарата.
Исследование эффективности
Показаниями для назначения препаратов являются:
- сокращение сроков продолжительности нейтропении после цитотоксической химиотерапии по поводу злокачественных заболеваний или миелобластивной терапии с последующей пересадкой костного мозга;
- мобилизация клеток предшественников периферических клеток крови;
- лечение врожденных, циклической или идиопатической нейтропений;
- лечение нейтропении, которая развилась на фоне ВИЧ-инфекции.
Режим дозирования препарата зависит от заболевания.
Наиболее подходящей клинической моделью для доказательства подобия биоаналогичного (биоподобного) и оригинального (референтного) препаратов является предупреждение тяжелой нейтропении, которая развивается после цитостатической химиотерапии, на однородной группе больных (по типу опухоли, предыдущей химиотерапии, запланированной химиотерапии и стадии болезни). В исследование должны быть включены больные, которым планируется проведение химиотерапии, вызывающей развитие тяжелой нейтропении. При использовании химиотерапии с заранее известной частотой и продолжительностью тяжелой нейтропении достаточно проведения исследования в двух сравниваемых группах. При использовании других схем химиотерапии может потребоваться исследование в трех группах, включая контрольную группу с использованием плацебо.
Основным показателем эффективности является продолжительность тяжелой нейтропении при уровне абсолютного количества нейтрофилов ниже 0,5 x 109/л, допустимый предел отклонений которого должен быть обоснован. Вторичными показателями эффективности являются частота появления фебрильной нейтропении, развитие инфекционного процесса и зависимость клинических проявлений от дозы рГ-КСФ. Главный акцент следует сделать на первом цикле проведения химиотерапии.
Доказательство подобия в клинических исследованиях на модели нейтропении, вызванной химиотерапией, позволяет экстраполировать результаты исследования эффективности на другие показания к применению, указанные в инструкции для оригинального (референтного) препарата, при условии, что они имеют аналогичный механизм действия.
При достаточном обосновании для демонстрации сходства могут быть использованы другие альтернативные модели исследования, включая изучение фармакодинамических свойств на здоровых добровольцах. Для этого необходимо представить научное обоснование выбора модели, дизайна исследования, продолжительности исследования, выбора доз, показателей и конечных точек исследования, эффективности и фармакодинамики и пределов сопоставимости.
Исследование безопасности
Данные по безопасности необходимо получать на группе пациентов, которым препарат вводился многократно, предпочтительно в ходе сравнительного клинического исследования. Суммарная экспозиция должна соответствовать экспозиции при обычном химиотерапевтическом лечении, содержащем несколько циклов. Общая продолжительность периода последующего наблюдения должна составлять не менее 6 месяцев. Числа пациентов должно быть достаточно для оценки профиля нежелательных явлений (включая боли в костях) и отклонений в результатах лабораторных исследований. Данные об иммуногенности должны быть собраны в соответствии с принципами, описанными в главе 15.2 настоящих Правил.
4.3. План фармаконадзора
При регистрации биоаналогичного (биоподобного) препарата Г-КСФ необходимо представить план управления рисками (план фармаконадзора) в соответствии с правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией, и актами, входящими в право Союза. Основное внимание должно быть уделено изучению иммуногенности и выявлению возможных редких тяжелых нежелательных явлений в первую очередь среди больных, получающих препарат длительно, и пациентов, у которых отмечается снижение эффективности препарата при проведении мобилизации клеток предшественников гемопоэза.
Глава 15.6. Доклинические и клинические исследования
биоаналогичных (биоподобных) лекарственных препаратов
на основе гепаринов низкой молекулярной массы
1. Вступление
Гепарин является высоко сульфированным и гетерогенным членом семейства гликозаминогликановых углеводов, состоящих из различных дисахаридных единиц. К наиболее распространенным дисахарам относятся: 2-O-сульфат 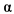 -L-идуроновой кислоты и 6-O-сульфат, N-сульфат
-L-идуроновой кислоты и 6-O-сульфат, N-сульфат 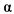 -D-глюкозамин и IdoA (2S)-GlcNS (6S). Эндогенный гепарин вырабатывается в гранулах тучных клеток (лаброцитов) и обладает самой высокой плотностью отрицательного заряда среди всех биологических молекул. Для изготовления препаратов гепаринов в основном используют гепарин, получаемый из слизистой оболочки кишечника свиней.
-D-глюкозамин и IdoA (2S)-GlcNS (6S). Эндогенный гепарин вырабатывается в гранулах тучных клеток (лаброцитов) и обладает самой высокой плотностью отрицательного заряда среди всех биологических молекул. Для изготовления препаратов гепаринов в основном используют гепарин, получаемый из слизистой оболочки кишечника свиней.
Гепарин угнетает образование нескольких сериновых протеаз системы свертывания крови посредством активации антитромбина. Основную роль в связывании гепарина с антитромбином играет пентасахаридная последовательность, содержащая 3-O-сульфатный глюкозаминовый остаток. После связывания с антитромбином гепарин вызывает конформационное изменение его молекулы, что приводит к активации области, ответственной за ингибирование активированных факторов свертывания. Кроме того, гепарин выступает в роли катализатора, связывая ингибитор и активированные протеазы серина (например, факторы IXa и XIa). Это свойство гепарина зависит главным образом от числа моносахаридов в молекуле гепарина.
Молекулы гепарина, содержащие менее 18 моносахаридов, не катализируют ингибирование тромбина, но угнетают действие фактора свертывания Xa. После того как сериновые протеазы начинают действовать на специфичную пептидную связь Arg-Ser (аргинин-серин) активного центра молекулы антитромбина, гепарин повышает скорость реакции между тромбином и антитромбином как минимум в тысячу раз, с образованием стабильного комплекса 1:1. Гепарин взаимодействует и с другими структурами плазмы и клеток, но в сравнении с ингибированием процесса свертывания крови их клиническое значение изучено недостаточно.
Гепарин вводят парентерально, поскольку при пероральном приеме он разрушается. Его можно вводить внутривенно, внутриартериально или подкожно, внутримышечных инъекций следует избегать из-за риска гематом.
Низкомолекулярные гепарины получают из нефракционированного гепарина в процессе химической или ферментативной деполимеризации. Исходным материалом для получения низкомолекулярных гепаринов является вещество биологического происхождения, а характеристики фармацевтической субстанции определяются производственным процессом.
Сложность состава низкомолекулярных гепаринов обусловлена преимущественно особенностями исходного материала (нефракционированный гепарин, выделенный из слизистой оболочки кишечника свиней или других тканей животных), процессами выделения, фракционирования и производства.
Существует несколько современных методов для физико-химической характеристики препаратов низкомолекулярных гепаринов. Тем не менее в настоящее время не известно, как именно множественные различия в полисахаридах влияют на клинические эффекты, связанные с эффективностью и безопасностью низкомолекулярных гепаринов.
Конкретный препарат низкомолекулярного гепарина отличается от нефракционированного гепарина и от других низкомолекулярных гепаринов по фармакокинетическим и фармакодинамическим свойствам. В результате процесса деполимеризации молекула действующего вещества содержит в основном цепочки, состоящие менее чем из 18 моносахаров. Подобное уменьшение размера молекулы сопровождается снижением ингибирующей активности тромбина в сравнении со стандартным гепарином и повышением ингибирующей активности фактора свертывания крови Xa.
Из-за сложности определения низкомолекулярных гепаринов в крови невозможно оценить фармакокинетические свойства препаратов на основе гепаринов. Однако оценить всасывание и элиминацию низкомолекулярных гепаринов можно с использованием таких фармакодинамических тестов, как оценка анти-Xa и анти-IIa активности.
Существует несколько зарегистрированных низкомолекулярных гепаринов, которые различаются между собой по исходному сырью, производственному процессу, фармакокинетическим (фармакодинамическим) свойствам и терапевтическим показаниям, включающим лечение и профилактику тромбоза глубоких вен, а также профилактику осложнений острого коронарного синдрома (нестабильная стенокардия, инфаркт миокарда как с подъемом сегмента ST, так и без него).
К наиболее частым нежелательным реакциям при применении гепаринов относятся кровотечения, а к наиболее серьезной нежелательной реакции относится редко наблюдаемая гепарин-индуцированная тромбоцитопения II типа. Данное осложнение развивается под влиянием образования антител к новым антигенам, которые образуются при формировании комплекса, содержащего тромбоцитарный фактор 4 (PF4) и гепарин. Связывание антител с новым антигеном (комплекс тромбоцитарный фактор 4 (PF4) и гепарин) вызывает активацию тромбоцитов с последующим образованием тромбогенных микроагрегатов тромбоцитов. У пациентов с тромбоцитопенией повышен риск артериальных и венозных тромбоэмболических осложнений (гепарин-индуцированная тромбоцитопения и тромбоз). Риск развития этих нежелательных реакций является более низким в сравнении с применением нефракционированного гепарина, но при назначении препаратов низкомолекулярного гепарина необходимо регулярно контролировать содержание тромбоцитов у всех пациентов, а у тех пациентов, у которых выявлена тромбоцитопения или тромбоэмболические осложнения, необходимо определение антител к комплексу PF4-гепарин.
Следует отметить, что гетерогенность низкомолекулярных гепаринов очень велика. При этом влияние структуры действующего вещества на механизм действия препарата до конца не изучено, а фармакодинамические маркеры анти-FXa и FIIa активности не позволяют в полной мере охарактеризовать (спрогнозировать) терапевтическую эффективность. Поэтому при проведении клинических исследований, как правило, необходимо охарактеризовать те моменты, которые не получили полную оценку в процессе изучения физико-химических и биологических свойств.
Настоящая глава применяется в дополнение к требованиям для демонстрации подобия (сходства) двух препаратов низкомолекулярного гепарина. Требования настоящей главы необходимо рассматривать совместно с другими соответствующими требованиями и актами, входящими в право Союза.
2. Сфера применения
Настоящая препарат-специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих низкомолекулярный гепарин.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Доклинические исследования
Перед началом клинических исследований должны быть проведены доклинические исследования. Доклинические исследования носят сравнительный характер и направлены на выявление различий между фармакотоксикологическим действием биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) низкомолекулярного гепарина, а не на изучение ответа на препарат как такового. Выбор подхода должен быть полностью обоснован в доклиническом обзоре.
Исследование фармакодинамики
Исследование in vitro
Для сопоставления разницы в активности подобного лекарственного препарата и оригинального (референтного) низкомолекулярного гепарина должны быть представлены данные нескольких сравнительных биологических анализов (на основании современных данных о клинически значимом фармакодинамическом влиянии низкомолекулярных гепаринов, включая как минимум оценку анти-Xa и анти-IIa активности). По возможности для измерения активности следует использовать стандартизированные методы (например, в соответствии с Фармакопеей Союза). Такие данные могут быть получены ранее в процессе изучения качества препарата.
Исследование in vivo
Если при характеристике физико-химических и биологических свойств с использованием высокочувствительных современных методов установлена высокая степень сходства биоаналогичного (биоподобного) и оригинального (референтного) препаратов, не требуется проведения исследований in vivo как части изучения сопоставимости. В остальных случаях in vivo сравнительное количественное изучение фармакодинамической активности биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярных гепаринов включает:
использование фармакодинамической модели in vivo, разработанной с учетом самых современных данных о клинически значимых фармакодинамических эффектах низкомолекулярных гепаринов, в которую включена по меньшей мере оценка анти-Xa и анти-IIa активности, а также оценка степени высвобождения ингибитора пути тканевого фактора, и (или)
использование подходящей модели либо венозного, либо артериального тромбоза на животных, в соответствии с клиническими показаниями.
Исследование токсичности
Как правило, изучение токсичности при повторных введениях одной дозы не требуется.
Если в состав препарата включено новое или малоизученное вспомогательное вещество, необходимо проведение дополнительных исследований токсичности.
Проведение сравнительных исследований неспецифической токсичности только для оценки установленных различий в составе примесей не рекомендуется. При разработке продукта a priori исходят из концепции его высокой степени подобия оригинальному (референтному) препарату, что должно быть подтверждено физико-химическими методами исследования. Поэтому наилучшей стратегией для снижения рисков, обусловленных примесями (например, белками) является их сведение к минимуму в соответствии с требованиями фармакопейной статьи (монографии).
Исследования фармакологической безопасности, репродуктивной токсичности не являются обязательными при сравнительном исследовании биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярных гепаринов. Исследования местной переносимости не проводятся, если в состав препарата не включены вспомогательные вещества, для которых не имеется достаточного документально подтвержденного опыта использования при данном пути введения лекарственного препарата или же он ограничен. Если выполнялись иные исследования in vivo, то оценка местной переносимости может быть проведена как их часть.
5. Клинические исследования
Исследование фармакокинетики и фармакодинамики
Гетерогенность низкомолекулярных гепаринов не позволяет проведение обычного исследования фармакокинетических свойств. Поэтому оценка всасывания и элиминации низкомолекулярных гепаринов проводится при изучении фармакодинамических свойств по показателям (включая анти-Xa и анти-IIa), которые могут быть использованы в качестве суррогатных маркеров концентрации препарата. При этом могут быть использованы и другие показатели, такие как активность ингибитора пути тканевого фактора и соотношение активности анти-Xa и анти-IIa факторов. Оценка данных показателей позволит в значительной степени охарактеризовать полисахаридный профиль препарата. Изучение подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов по данным показателям (ФК-профили и ФД-профили) проводят в рандомизированном перекрестном исследовании в двух группах здоровых добровольцев при подкожном введении препаратов. Если оригинальный (референтный) препарат кроме подкожного пути введения зарегистрирован еще и для внутривенного или внутриартериального пути введения, то необходимо проведение дополнительного сравнительного исследования для данных путей введения.
Выбранная доза должна соответствовать крутой части кривой зависимости "доза - эффект" и находиться в рекомендуемом для разных показаний диапазоне доз. Пределы эквивалентности также должны быть определены и должным образом обоснованы заранее.
Исследование эффективности
Как правило, для демонстрации сходства биоаналогичного (биоподобного) и оригинального (референтного) препаратов необходимо сравнительное изучение клинической эффективности. Сравнительные исследования эффективности могут не проводиться только в том случае, если биоаналогичный (биоподобный) и оригинальный (референтный) низкомолекулярный гепарины могут быть достоверно сопоставлены по своим физико-химическим характеристикам, биологической активности (силе действия) и характерному фармакодинамическому профилю установленным с применением современных высокочувствительных и специфичных методов исследования. Следует принимать во внимание, что это возможно в исключительном случае, при условии, что в регистрационном досье представлены данные значительного количества проведенных подтверждающих химико-аналитических и биоаналитических исследований.
Терапевтическая эквивалентность должна быть подтверждена в клиническом исследовании достаточной статистической мощности, рандомизированном, двойном слепом, проводимом в параллельных группах. Теоретически это можно осуществить при применении препарата для профилактики венозной или артериальной тромбоэмболии или при лечении венозной тромбоэмболии. При этом должна быть выбрана наиболее чувствительная модель для выявления различий эффективности между новым низкомолекулярным гепарином и оригинальным (референтным) лекарственным препаратом.
Наиболее высокий показатель распространения венозной тромбоэмболии отмечается у хирургических больных, кроме того, подавляющее большинство опубликованных исследований проводилось с участием хирургических пациентов с высоким риском развития венозной тромбоэмболии, что особенно характерно для пациентов после полного эндопротезирования тазобедренного или коленного сустава. Таким образом, данная популяция больных наиболее полно изучена и накоплен большой объем данных, касающихся данных операции, продолжительности исследования и рисков развития кровотечений.
Учитывая это, оценку сравнительной эффективности рекомендуется проводить при применении препарата для профилактики венозной тромбоэмболии у пациентов, которым были проведены операции с высоким риском развития венозных тромбоэмболических осложнений. Предпочтительно проведение исследований с участием больных, перенесших обширное ортопедическое оперативное вмешательство, например, на тазобедренном суставе. В клинические исследования рекомендуется включать достаточное количество больных с переломом шейки бедра, поскольку они обладают и высоким риском тромбозов, и высоким риском оперативного кровотечения. Доза препарата должна соответствовать дозе, указанной в инструкции для применения оригинального (референтного) препарата для профилактики осложнений венозной тромбоэмболии.
При этом для выявления возможных различий эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов сравниваемые группы больных должны быть максимально однородными.
В качестве комбинированной конечной точки эффективности профилактики венозных тромбоэмболических осложнений можно использовать показатель частоты развития проксимального тромбоза глубоких вен, тромбоэмболии легочной артерии и летальных исходов, обусловленных венозной тромбоэмболией. Для сравнительной оценки двух препаратов можно использовать показатель суммарной точки, которая рассчитывается как сумма общего числа тромбоэмболических осложнений (тромбоэмболия легочной артерии и венозные тромбоэмболии, количество летальных исходов, обусловленных тромбозом глубоких вен). Решение о наличии венозной тромбоэмболии должно быть вынесено центральной независимой комиссией экспертов, на основании анализа "ослепленных" данных.
Для проведения сравнительных исследований необходимо определить статистически и клинически обоснованные допустимые пределы отклонения показателей. Исследование проводится для доказательства подобия (сходства) эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов на основании одной из указанных выше конечных точек.
Для оценки конечной точки необходимо использовать современные диагностические методики. Если для определения проксимального тромбоза глубоких вен с высокой специфичностью и чувствительностью можно использовать УЗИ, то выявить дистальный тромбоз глубоких вен можно только с помощью двусторонней венографии. Поэтому использование данных методик является обязательным для определения конечных точек.
Для демонстрации подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов необходимо использовать соответствующие конечные точки, такие как количество случаев тромбоза глубоких вен, тромбоэмболии легочной артерии и летальных исходов.
Оценка первичной конечной точки проводится в момент появления симптомов, указывающих на венозную тромбоэмболию, или у пациентов без симптоматики в конце лечения. Наблюдение за больными должно проводиться не менее 60 дней, чтобы можно было обнаружить поздние тромботические осложнения.
Исследование безопасности
Даже если сделан вывод об аналогичной эффективности на основании сопоставления данных по физико-химическим свойствам, биологической активности (силе действия) и характерным фармакодинамическим профилям, до представления заявления на регистрацию биоаналогичного (биоподобного) низкомолекулярного гепарина необходимо проведение сравнительной оценки его безопасности у человека.
Для регистрации биоаналогичного (биоподобного) низкомолекулярного гепарина может быть достаточно данных о сравнительной безопасности, которые были получены в рамках клинических исследований эффективности в предрегистрационный период. Следует дать подробную сравнительную оценку нежелательных реакций по типу, частоте и тяжести между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами. Обширные и клинически значимые необширные кровотечения должны быть зарегистрированы и описаны. При этом необходимо использовать клинически релевантную классификацию кровотечений. Подобно оценке эффективности, решение о геморрагических явлениях должно выноситься центральным комитетом независимых экспертов на основании ослепленных данных и использовании заранее установленных критериев. Рекомендуется контролировать функцию печени.
При изучении безопасности необходимо продемонстрировать достаточным числом повторных анализов, что иммуногенность биоаналогичного (биоподобного) препарата не превышает иммуногенность оригинального (референтного) препарата. Для выявления гепарин-индуцированной иммунной тромбоцитопении (тип II) на протяжении всего исследования у пациентов с тромбоцитопенией и (или) тромбоэмболией необходимо определять количество тромбоцитов с использованием адекватных диагностических текстов (включая определение антител к комплексу PF4-гепарин). Определение антител у всех пациентов не имеет смысла, так как частота гепарин-индуцированной иммунной тромбоцитопении (тип II) очень низкая (определяется в < 0,1% случаев) и она вряд ли может быть выявлена на этапе предрегистрационного исследования.
6. План фармаконадзора
При регистрации биоаналогичного (биоподобного) препарата необходимо представить план по управлению рисками в соответствии с правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией и иными актами, входящими в право Союза. В плане должны быть отражены выявленные и потенциальные риски применения оригинального (референтного) препарата в соответствии с инструкцией по применению оригинального (референтного) препарата, а также мероприятия по отслеживанию параметров безопасности применения для соответствующих показаний оригинального (референтного) препарата, на которые проводилась экстраполяция результатов исследований по другим показаниям. Кроме того, необходимо представить план управления рисками, связанными с серьезными нежелательными явлениями при приеме препаратов низкомолекулярных гепаринов (например, гепарин-индуцированная тромбоцитопения II типа, анафилактические и анафилактоидные реакции).
7. Экстраполяция показаний
Демонстрация подобной (сходной) эффективности и безопасности биоаналогичным (биоподобным) и оригинальным (референтным) препаратами в популяции хирургических больных с высоким риском венозной тромбоэмболии при достаточном обосновании позволяет экстраполировать полученные результаты исследований на другие показания, указанные для оригинального (референтного) препарата.
Глава 15.7. Доклиническая и клиническая разработка
биоаналогичных (биоподобных) лекарственных препаратов,
содержащих рекомбинантный инсулин и аналоги инсулина
Настоящая глава устанавливает доклинические и клинические требования к лекарственным препаратам, содержащим рекомбинантный инсулин, включая инсулин человека и аналоги инсулина человека (собирательно называемые инсулином), заявленным в качестве аналогичных другому зарегистрированному лекарственному препарату (оригинальному (референтному) лекарственному препарату).
В доклиническом разделе рассматриваются требования к in vitro фармакодинамическим исследованиям и ситуациям, которые могут потребовать дополнительной токсикологической оценки in vivo. В клиническом разделе рассматриваются требования к фармакокинетическим, фармакодинамическим исследованиям и исследованиям безопасности, а также плану управления рисками.
В сферу применения были включены препараты инсулина средней продолжительности и длительного действия, а также аналоги инсулина. В отношении доклинических исследований in vivo вводится подход, основанный на рисках, даются более подробные рекомендации по дизайну, исследуемой популяции, дозам инсулина и конечным точкам инсулинового клэмп-исследования. Кроме того, подробно охарактеризованы требования к исследованиям безопасности, а также включены обязательные условия, при выполнении которых возможно непроведение такого исследования.
1. Введение
Регистрационное досье рекомбинантного инсулина человека или его аналога, заявленного в качестве аналогичного зарегистрированному оригинальному (референтному) лекарственному препарату, должно содержать подтверждение биоаналогичности заявленного препарата такому оригинальному (референтному) лекарственному препарату.
Инсулин человека - негликозилированной содержащий дисульфидную связь гетеродимер, состоящий из 51 аминокислоты. Аналоги инсулина отличаются от инсулина человека заменой аминокислот или другими химическими модификациями, такими как включение цепи жирной кислоты в молекулу. Препараты инсулина отличаются преимущественно по кинетическому и (или) фармакодинамическому профилям. Как правило, выделяют препараты ультракороткого (действие развивается быстрее, чем у растворимого инсулина человека), короткого (например, растворимый инсулин человека), средней продолжительности (например, инсулин-изофан человека = инсулин НПХ) и длительного действия (инсулины с профилями действия, существенно превышающими таковой инсулин НПХ) и применяются в монотерапии в составе экстемпоральной смеси или комбинированных препаратов инсулина ультракороткого (короткого) действия и инсулина средней продолжительности (длительного) (двухфазного) действия в различных соотношениях.
Для детального установления характеристик первичной, вторичной и третичной структур молекулы рекомбинантного инсулина, а также его аффинности с рецептором и биологической активности in vitro и in vivo доступны подходящие физико-химические и биологические методы. Необходимо уделить внимание родственным соединениям и родственным примесям, а также производственным примесям, в особенности дезамидо-формам, гликозилированным формам и прочим формам, которые могут быть обусловлены экспрессирующей системой или проистекать на этапах преобразования с целью удаления C-пептида и восстановления третичной структуры.
Доступные в настоящее время инсулины вводят подкожно и внутривенно. Действие инсулина преимущественно опосредовано стимуляцией инсулинового рецептора, однако инсулин также является слабым естественным лигандом рецептора инсулиноподобного фактора роста-1 (ИФР-1).
К инсулину часто вырабатываются антитела, преимущественно перекрестно реагирующие. Они, как правило, не сказываются на эффективности или безопасности. Необходимо оценить потенциал выработки антител к препарату и примесям. Возможные факторы риска развития иммунного ответа, обусловленные пациентом, неизвестны.
2. Сфера применения
Настоящая препаратспецифичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих рекомбинантный инсулин человека.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Доклинические исследования
Перед началом клинической разработки необходимо провести доклинические исследования. Эти исследования должны носить сравнительный характер и быть спланированы с целью достижения необходимой чувствительности для выявления значимых различий в ответ на биоаналогичный (биоподобный) лекарственный препарат и оригинальный (референтный) лекарственный препарат, а не оценивать ответ per se. Принятый подход необходимо всесторонне обосновать в доклиническом обзоре регистрационного досье.
Фармакодинамические исследования
Исследования in vitro
В целях оценки любых различий в свойствах между биоаналогичным (биоподобным) и референтным лекарственными препаратами необходимо провести сравнительные in vitro испытания на связывание с рецептором, а также испытания на последующую биологическую активность. Частично эти данные могут быть доступны по результатам испытаний, проведенных с целью определения активности при оценке физико-химических характеристик. Необходимо подтвердить чувствительность испытаний при изучении сопоставимости для выявления любых значимых различий и то, что эксперименты проведены с достаточным числом повторностей, разведений или временных точек на кривой с целью правильной характеристики зависимости "концентрация - эффект" или "время - эффект". Биоаналогичный (биоподобный) и оригинальный (референтный) лекарственные препараты необходимо сравнивать параллельно в одном эксперименте. Все испытания должны включать надлежащие контроли, подтверждающие валидность и пригодность метода.
Необходимо подтвердить сопоставимость связывания с обоими рецепторами инсулина человека (ИР-A и ИР-B), в том числе кинетику включения - выключения. С этой целью возможно использование клеток искусственно экспрессирующих ИР-A или ИР-B соответственно. При использовании клеток, эндогенно экспрессирующих ИР-A или ИР-B, необходимо подтвердить то, что действительно присутствует лишь один рецептор. Иначе возникают затруднения в интерпретации результатов связывания. При использовании другого современного метода изучения связывания необходимо обосновать выбор такого метода.
Биологическую активность необходимо сравнить на двух уровнях: (1) аутофосфорилирование рецептора и (2) метаболическая активность. В целом митогенная активность, опосредуемая стимуляцией рецептора ИФР-1, может быть не значима для инсулина человека и большинства аналогов инсулина. Однако, если применимо, допускается изучить сравнительное связывание с рецептором ИФР-1 и функциональную активность, чтобы охватить этот потенциальный токсикологический эффект. В отношении аутофосфорилирования рецепторов необходимо обеспечить, чтобы динамический диапазон используемого в испытании метода обнаружения не был слишком узким, поскольку это снизит возможность обнаружения значимых различий в степени аутофосфорилирования рецептора. Доступны различные методы изучения метаболической активности, включая методы определения образования гликогена, липогенеза, ингибирования стимулированного липолиза, а также транспорта глюкозы. Эти процессы можно изучить на различных клетках. В целях подтверждения сопоставимости необходимо использовать по меньшей мере три различных метода изучения метаболической активности. Данные должны давать четкое видение соотношения агонистических свойств биоаналогичного (биоподобного) и референтного лекарственных препаратов в отношении инсулинового рецептора. Выбор методов изучения метаболической активности необходимо обосновать в соответствии с указанными критериями.
Исследования in vivo
Сравнительные in vivo исследования фармакодинамических эффектов скорее всего будут недостаточно чувствительны для обнаружения различий, не выявленных в in vitro испытаниях, в связи с чем включать их в программу изучения сопоставимости не требуется.
Токсикологические исследования
Отдельные исследования токсичности при многократном введении не требуются. В определенных случаях, например, при использовании новых вспомогательных веществ, придерживаясь подхода, основанного на рисках, следует определить необходимость проведения дополнительных токсикологических исследований в соответствии с главой 15.2 настоящих Правил.
При доклиническом исследовании биоаналогичного (биоподобного) лекарственного препарата, содержащего инсулин или аналоги инсулина, проведение исследований фармакологической безопасности, репродуктивной токсичности и канцерогенности не требуется. Проведение исследований местной переносимости не требуется, если только не использованы вспомогательные вещества, в отношении которых отсутствует опыт (или он ограничен) их применения при намеченном пути введения.
5. Клинические исследования
Исследования фармакологии
В дополнение к результатам доказательства сходства (подобия) физико-химических и функциональных характеристик подтверждение сходства (подобия) фармакокинетического и фармакодинамического профилей является основой доказательства сходной (подобной) эффективности биоаналогичного (биоподобного) и оригинального (референтного) инсулина. Для достижения этой цели наиболее подходящими считаются перекрестные, предпочтительно двойные слепые гиперинсулинемические эугликемические клэмп-исследования при однократном подкожном введении биоаналогичного (биоподобного) и оригинального (референтного) инсулина. Во избежание эффектов переноса при определении отмывочной фазы необходимо учитывать продолжительность действия исследуемого препарата инсулина. Профили "время - концентрация" и "время - действие" предпочтительно изучать одновременно (в одном клэмп-исследовании). Дополнительные фармакологические исследования при внутривенном введении не требуются.
Исследуемая популяция
В целях наилучшего выявления различий, обусловленных препаратами, исследуемая популяция должна быть однородной и инсулин-чувствительной, ею могут быть здоровые добровольцы или пациенты с сахарным диабетом 1-го типа с нормальной массой тела.
Помимо большей доступности здоровые добровольцы проявляют меньшую внутрииндивидуальную вариабельность по сравнению с пациентами с сахарным диабетом 1-го типа (СД1Т), однако их недостатком является наличие эндогенного инсулина, который, не считая некоторых аналогов инсулина, невозможно с помощью доступных методов отличить от экзогенно вводимого инсулина. Возможны использование методов подавления секции эндогенного инсулина или коррекция значений сывороточной концентрации инсулина на вычисленную концентрацию эндогенного инсулина.
В целях обеспечения отсутствия значимой остаточной секреции эндогенного инсулина необходимо осуществлять скрининг содержания C-пептида у пациентов с СД1Т, включаемых в клэмп-исследования. В целях достижения сопоставимых базовых условий во всех экспериментах необходимо обеспечить стабильное и сопоставимое базовое содержание глюкозы в крови и концентрации инсулина в течение некоторого времени (в идеале в течение одного часа) до начала исследования, что может вызвать большие затруднения у пациентов с СД1Т, чем у здоровых субъектов.
В целях сравнения инсулинов короткого или средней продолжительности действия в клэмп-исследования допускается включать как здоровых субъектов, так и пациентов с СД1Т, тогда как для сравнения инсулинов длительного действия предпочтительно исследовать пациентов с СД1Т.
Чувствительность к инсулину у женщин может варьировать в течение менструального цикла. На сегодняшний день не ясно, может ли это повлиять на результаты исследования. В связи с этим в исследования предпочтительно включать только мужчин.
Инсулиновый клэмп ("инсулиновые тиски")
По общему мнению, эугликемический гиперинсулинемический клэмп-метод является наилучшим из доступных методов определения действия инсулина. В таких клэмп-экспериментах увеличивают плазменную концентрацию инсулина (например, за счет его подкожного введения), а содержание глюкозы в крови поддерживают ("зажимают в тиски") на заданном уровне с помощью регулирования введения глюкозы.
Для поддержания содержания глюкозы в крови существуют различные клэмп-методы и алгоритмы обратной связи. Клэмп-исследования можно проводить вручную или с помощью автоматизированной процедуры. Оба подхода требуют большого опыта. Однако оба метода дают схожие и воспроизводимые результаты, если отсутствует быстрое изменение потребности в глюкозе, которое может быть вовремя не распознано в зависимости от длительности интервалов между определением содержания глюкозы в крови в ходе ручного клэмп-метода. Настоятельно рекомендуется использовать двойной слепой дизайн, особенно при ручном методе, который больше подвержен систематическим ошибкам со стороны исследователя по сравнению с автоматизированным клэмп-исследованием. При невозможности этого необходимо прибегнуть к другим способам эффективного снижения потенциальных систематических ошибок со стороны исследователя.
В целях снижения вариабельности условия эксперимента в сравнительном клэмп-исследовании необходимо как можно больше стандартизировать. Во избежание искажающего влияния на результаты исследуемых субъектов следует включать в клэмп-эксперименты после ночного голодания (как правило, в течение 10 - 12 часов), и они должны оставаться голодными в ходе всего исследования. У пациентов с сахарным диабетом необходимо минимизировать эффекты переноса, обусловленные введением инсулина до исследования. В идеале целевого содержания глюкозы в клэмп-эксперименте необходимо достичь по меньшей мере за час до экспериментального введения инсулина без введения глюкозы в течение этого последнего часа. Необходимо обеспечить стандартизацию клэмп-метода и факторов, влияющих на чувствительность к инсулину, таких как время суток, физическая активность, прием пищи и рацион, отказ от алкоголя, кофеинсодержащих напитков, курения, препаратов, за исключением исследуемых, и отсутствие сопутствующих заболеваний (в том числе инфекций) и психического стресса. В исследовательском центре субъектам следует позволить адаптироваться к экспериментальным условиям, чтобы достичь сопоставимого метаболического статуса, они должны находиться в спокойном окружении и избегать физической активности в ходе всего исследования. Эти требования свидетельствуют о важности мельчайших деталей.
При включении в клэмп-исследования здоровых добровольцев выработка эндогенного инсулина у них может искажать ФК-параметры и (или) ФД-параметры. Существуют специфические методы, способные различать некоторые аналоги инсулина от эндогенного инсулина. При наличии целесообразно использовать эти методы. В целях оценки прандиального инсулина считается, что болюсное введение инсулина будет в достаточной степени подавлять эндогенный инсулин в течение клэмп-эксперимента. Секрецию эндогенного инсулина можно, как правило, в достаточной степени подавить, удерживая концентрацию глюкозы в крови ниже ее содержания у субъекта натощак. В качестве альтернативы возможно введение первичной дозы инсулина ультракороткого или короткого действия с последующим поддержанием базовой скорости (например, 0,10 - 0,15 мЕД/мин/кг), однако показано, что дополнительная инфузия базального инсулина искажает поздний глюкодинамический профиль инсулина НПХ и, возможно, даже в большей степени препаратов инсулина длительного действия, завышая эффект исследуемых инсулинов. В целях максимального подавления секреции эндогенного инсулина, глюкагона и гормона роста в ходе клэмп-исследований прибегали к введению соматостатина, но в связи с низкой переносимостью его широкое применение не рекомендуется. Кроме того, следует отметить, что соматостатин снижает клиренс инсулина, искусственно увеличивая, таким образом, продолжительность его действия. В клэмп-исследованиях с участием здоровых добровольцев необходимо всегда определять концентрацию C-пептида параллельно с концентрацией инсулина на протяжении всего эксперимента с целью оценки степени и постоянства подавления секреции эндогенного инсулина. В отсутствие подавления секреции инсулина можно прибегнуть к методам коррекции по содержанию C-пептида. Независимо от используемого метода в целях обеспечения сопоставимых экспериментальных условий он должен быть обоснован и одинаков во всех клэмп-исследованиях.
Инсулин в клэмп-исследованиях наиболее часто вводят в дозах 0,2 - 0,3 ЕД/кг массы тела - инсулины ультракороткого и короткого действия, 0,3 - 0,4 ЕД/кг массы тела - инсулины средней продолжительности действия и 0,4 - 0,6 ЕД/кг массы тела - инсулины длительного действия. Дозы в верхнем диапазоне, как правило, дают более надежный ФД-ответ, снижая тем самым ФД-вариабельность. Ожидается, что достигаемая степень гиперинсулинемии будет находиться на крутой части кривой "доза - эффект" инсулина, то есть можно ожидать высокую чувствительность в выявлении потенциальных различий в профилях "время - действие" двух инсулинов. В целях снижения вариабельности необходимо стандартизировать место и технику введения.
Концентрация глюкозы в крови у здоровых добровольцев обычно удерживается ниже (например, на 0,3 ммоль/л (5 мг/дл), или 10 процентов) содержания глюкозы у субъекта натощак или при 4,4 - 5,6 ммоль/л (80 - 100 мг/дл). У пациентов с СД1Т, содержание глюкозы в крови обычно удерживается при 5,6 ммоль/л (100 мг/дл). Необходимо заранее указать допустимые отклонения в содержании сахара крови от этого значения в ходе клэмп-исследования. Необходимо избегать снижения концентрации глюкозы ниже 3,3 ммоль/л (60 мг/дл), поскольку это может привести к стимуляции выделения контр-регуляторных гормонов (адреналина, глюкагона, кортизола, гормона роста) для повышения концентрации глюкозы в крови и привести к быстрому и выраженному снижению чувствительности к инсулину, влияя на оцениваемый профиль "время - активность" исследуемого препарата инсулина.
При определении продолжительности клэмп-исследований необходимо учитывать известную продолжительность действия препарата исследуемого инсулина и его дозозависимость. Продолжительность действия в "глюкозных тисках" может быть задана как время от момента введения инсулина до восстановления скорости введения глюкозы (СВГ) до базового или заранее установленного значения (например, 0,5 мг/кг/мин) или у пациентов с сахарным диабетом - до концентрации глюкозы в крови, превышающей заранее установленный порог, например, 8,3 ммоль/л (150 мг/дл). Обычная продолжительность клэмп-эксперимента составляет 8 - 10 часов и 10 - 12 часов для инсулинов ультракороткого и короткого действия соответственно. Рекомендуемая продолжительность клэмп-исследования для препаратов инсулина средней продолжительности и длительного действия составляет по меньшей мере 24 часа.
Необходимо во всех случаях представлять обоснование выбора продолжительности клэмп-эксперимента, принимая во внимание известное влияние дозы инсулина и применения соматостатина (если применимо) на продолжительность действия инсулина и этнические различия в клиренсе инсулина.
Конечные точки и статистический анализ
Фармакокинетика
В качестве первичных конечных точек изучения инсулинов ультракороткого и короткого действия следует выбрать AUC(0-t) и Cmax, а в качестве вторичных конечных точек: 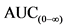 , частичные AUC (подобающие для соответствующего инсулина), tmax и t1/2.
, частичные AUC (подобающие для соответствующего инсулина), tmax и t1/2.
В качестве первичных конечных точек изучения инсулинов средней продолжительности действия следует выбрать 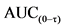 и Cmax, а в качестве вторичных конечных точек: AUC(0-t),
и Cmax, а в качестве вторичных конечных точек: AUC(0-t),  , подобающие частичные AUC, tmax и t1/2.
, подобающие частичные AUC, tmax и t1/2.
Инсулины длительного действия, как правило, проявляют плоский ФК-профиль. В связи с этим в некоторых случаях определение Cmax и tmax не всегда возможно и может не иметь смысла с клинической точки зрения. В таких случаях в качестве первичной конечной точки следует выбрать 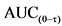 , а в качестве вторичных - частичные AUC, например,
, а в качестве вторичных - частичные AUC, например, 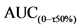 и
и 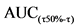 . По возможности следует определять t1/2.
. По возможности следует определять t1/2.
90 процентный доверительный интервал отношения первичных ФК-конечных точек исследуемого препарата к оригинальному (референтному) препарату должен укладываться в заранее выбранные границы эквивалентности. В отсутствие специальных для биологических лекарственных препаратов допустимых границ в целом и для инсулина в частности, в отсутствие обоснования иного следует выбрать стандартный диапазон биоэквивалентности, то есть 80 - 125 процентов. Если ожидается высокая вариабельность, в целях обоснования расширения допустимого диапазона следует воспользоваться повторным (репликативным) дизайном (например, трехпериодное перекрестное исследование с повторным введением препарата сравнения) в соответствии с правилами проведения исследований биоэквивалентности лекарственных препаратов в рамках Союза, утверждаемыми Комиссией.
Фармакодинамика (ФД)
Изменение СВГ во времени характеризует профиль "время - действие" препарата инсулина.
В качестве первичных конечных точек инсулинов ультракороткого и короткого действия, как правило, следует измерять СВГ-AUC(0-t) и СВГmax, СВГ-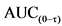 и СВГmax - для инсулинов средней продолжительности действия и СВГ-
и СВГmax - для инсулинов средней продолжительности действия и СВГ-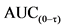 - для инсулинов длительного действия. К другим информативным фармакодинамическим конечным точкам относятся время до начала действия и tСВГmax - для инсулинов ультракороткого, короткого и средней продолжительности действия и частичные СВГAUC (информативные для соответствующего инсулина).
- для инсулинов длительного действия. К другим информативным фармакодинамическим конечным точкам относятся время до начала действия и tСВГmax - для инсулинов ультракороткого, короткого и средней продолжительности действия и частичные СВГAUC (информативные для соответствующего инсулина).
Если с помощью исчерпывающего аналитического установления характеристик и доклинических in vitro испытаний с использованием чувствительных ортогональных современных методов удается подтвердить высокую аналогичность физико-химических и функциональных характеристик биоаналогичного (биоподобного) и оригинального (референтного) инсулина, все СВГ-параметры можно отнести к вторичным точкам. Тем не менее ФД-данные должны всегда соотноситься с ФК-данными.
95 процентные доверительные интервалы отношения первичных ФД-параметров биоаналогичного (биоподобного) и оригинального (референтного) инсулина должны укладываться в заранее выбранные границы эквивалентности. При проведении исследования с повторным дизайном необходимо также документировать внутрииндивидуальную вариабельность ФД-конечных точек.
Качество инсулинового клэмп-исследования
Контроль концентрации глюкозы в крови ходе клэмп-исследования вызывает большие затруднения. В зависимости от интервалов осуществления замеров и алгоритма обратной связи и вследствие неизбежного запаздывания замеров между взятием образцов и коррекцией введения глюкозы и последующего запаздывания изменения концентрации глюкозы в крови в ответ на изменения СВГ значения концентрации глюкозы в крови, как правило, не соотносятся с точным целевым значением, а колеблются вокруг него. В связи с этим возникает вариация ("шум") в СВГ. Заявитель должен представить оценку качества проведения клэмп-исследования, например, в виде расчета средних значений, среднеквадратичного отклонения и коэффициента вариации концентрации глюкозы в крови. Необходимо проанализировать результаты и по возможности сравнить их с результатами, описанными в литературе. Необходимо также представить перечень индивидуальных клэмп-результатов. Шум в измерении СВГ в целях расчета СВГmax и временных параметров (например, tСВГmax) можно снизить с помощью математического моделирования. Необходимо заранее указать алгоритм коррекции СВГ и подтвердить корректность использованного метода сглаживания. В противоположность этому колебания (флуктуации) не сильно влияют на СВГ-AUC, поэтому ее можно рассчитать на основе несглаженных результатов измерения СВГ.
Особенности изучения препаратов
инсулина длительного действия
Препараты инсулина длительного действия предназначены для получения профиля "время - концентрация", который, насколько возможно, воспроизводит базальную физиологическую секрецию инсулина. При очень плоском ФК-профиле определение Cmax и tmax (инсулина и СВГ) может оказаться невозможным и не иметь смысла. Вследствие медленного снижения действия инсулина и неизбежной вариабельности СВГ, особенно в "хвостовой части" СВГ-кривой, может оказаться затруднительным определение продолжительности действия инсулина длительного действия, особенно у здоровых добровольцев вследствие искажающего влияния эндогенного инсулина. В связи с этим более пригодными для определения профиля "время - действие" инсулинов длительного действия являются пациенты с сахарным диабетом 1-го типа.
С другой стороны, сравнение хвостовой части профиля инсулин/СВГ инсулина длительного действия, вводимого, к примеру, один раз в сутки, может не иметь высокую клиническую значимость, поскольку остаточное содержание инсулина и действие инсулина от предыдущего введения, как правило, будет небольшим по сравнению с эффектом очередной дозы инсулина. По этой причине в качестве первичной ФК-конечной точки рекомендуется выбирать 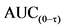 , а не AUC(0-t) (в соответствии с разделом "Конечные точки и статистический анализ" настоящей главы). Обязанностью заявителя является обоснование выбора исследуемой популяции и чувствительности экспериментальной модели и условий эксперимента для выявления значимых различий (при их наличии) между ФК-профилями и ФД-профилями биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов.
, а не AUC(0-t) (в соответствии с разделом "Конечные точки и статистический анализ" настоящей главы). Обязанностью заявителя является обоснование выбора исследуемой популяции и чувствительности экспериментальной модели и условий эксперимента для выявления значимых различий (при их наличии) между ФК-профилями и ФД-профилями биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов.
Несмотря на упомянутые выше ограничения и повышенную внутрииндивидуальную вариабельность инсулинов длительного действия по сравнению с инсулинами короткого действия, гиперинсулинемическое эугликемическое клэмп-исследование показало свою успешность как инструмент сравнения ФК-профилями и ФД-профилей зарегистрированных лекарственных препаратов инсулина длительного действия.
Требования к различным препаратам, содержащим одинаковую
активную фармацевтическую субстанцию
Если производитель биоаналогичного (биоподобного) лекарственного препарата разрабатывает другой препарат (например, препараты короткого действия, средней продолжительности действия и двухфазный, содержащие одинаковую активную фармацевтическую субстанцию), ФД-данные для всех этих препаратов не требуются. Для подтверждения аналогичной эффективности таких препаратов инсулина по отношению к соответствующим препаратам сравнения будет приемлема следующая программа:
1) подтверждение аналогичности ФК-профилей и ФД-профилей для препаратов растворимого инсулина;
2) подтверждение аналогичности ФК-профилей таких других препаратов инсулина по отношению к соответствующим оригинальным (референтным) лекарственным препаратам. Необходимо представить все ФД-данные, полученные в ходе ФК-исследований.
Клиническая эффективность
Проводить отдельные исследования эффективности не требуется, поскольку конечные точки, изучаемые в этих исследованиях (обычно это HbA1), считаются недостаточно чувствительными для выявления потенциальных клинически значимых различий между двумя инсулинами.
Клиническая безопасность
Исследования безопасности следует проводить с прицелом на иммуногенность. Исследования безопасности должны включать достаточное число пациентов с сахарным диабетом 1-го типа. Если включена смешанная популяция, необходима стратификация по типу сахарного диабета и наличию изначально присутствующих антиинсулиновых антител. Как правило, ослепление данных от участников исследования невыполнимо, однако по меньшей мере антиинсулиновые антитела следует определять слепым методом. Поскольку ожидается достаточно раннее образование антител, как правило, достаточно 6-месячного исследования, направленного на сравнение частоты образования и титров антител к исследуемому препарату и оригинальному (референтному) лекарственному препарату. С целью подтверждения не худшей иммуногенности достигать высокую мощность исследования нет необходимости. Тем не менее размер такого исследования должен однозначно исключить клинически значимое повышение иммуногенности. Необходимо изучить потенциальное влияние антиинсулиновых антител (при их обнаружении) на контроль гликемии, потребность в инсулине и безопасность, особенно местные и системные реакции гиперчувствительности.
Если в ходе исследования применяется фоновый инсулин (например, зарегистрированный прандиальный или базальный инсулин в дополнение к исследуемому инсулину), в ходе периода оценки изменять вид и режим применения фонового инсулина не допускается. Если производитель биоаналогичного (биоподобного) лекарственного препарата разрабатывает другие препараты, например, короткого, среднего действия и двухфазный препараты, содержащие одинаковый активный ингредиент, в исследование безопасности необходимо включить препарат с наибольшим ожидаемым иммуногенным потенциалом (изолированно или в комбинации с другими препаратами). Если препарат содержит вспомогательные вещества, в отношении которых отсутствует опыт применения или он ограничен, необходимо будет оценить безопасность и иммуногенность этого состава.
В определенных случаях допускается не проводить предрегистрационное исследование безопасности, предусматривающее оценку иммуногенности. Должны быть выполнены следующие условия: во-первых, должна быть убедительно показана биоаналогичность между биоаналогичным (биоподобным) и оригинальным (референтным) инсулином путем установления физико-химических и функциональных характеристик и сравнения их с помощью чувствительных, ортогональных современных аналитических методов, а также путем сравнения фармакокинетических и фармакодинамических профилей. Эти данные уже дают достаточную гарантию того, что можно ожидать одинаковую частоту возникновения нежелательных лекарственных реакций, которые опосредуются чрезмерными фармакологическими эффектами (например, гипогликемия). Во-вторых, профиль примесей и свойства вспомогательных веществ биоаналогичного (биоподобного) лекарственного препарата не должны давать повода для опасений. Необходимо во всех случаях представлять надлежащее научное обоснование отказа от проведения исследования безопасности (иммуногенности).
6. План фармаконадзора
В рамках процедуры регистрации заявитель должен представить план управления рисками в соответствии с правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией и актами, входящими в право Союза. В плане управления рисками биоаналогичного (биоподобного) лекарственного препарата необходимо всегда учитывать идентифицированные и потенциальные риски, обусловленные применением оригинального (референтного) лекарственного препарата. Кроме того, необходимо подробно проанализировать, как эти опасения относительно безопасности будут учтены в ходе пострегистрационного наблюдения.
7. Экстраполяция показаний
Подтверждение биоаналогичности, основанное на установлении физико-химических и функциональных характеристик, фармакокинетическом и (при необходимости) фармакодинамическом профилях и отсутствии вопросов с точки зрения безопасности при подкожном введении, позволяет провести экстраполяцию на внутривенное введение (при необходимости) и на другие показания и популяции пациентов, зарегистрированных для оригинального (референтного) лекарственного препарата.
8. Определения
Фармакокинетические параметры:
AUC(0-t) - площадь под кривой плазменной концентрации после введения и до завершения клэмп-эксперимента в момент t;
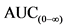 - площадь под кривой плазменной концентрации с экстраполяцией на бесконечность;
- площадь под кривой плазменной концентрации с экстраполяцией на бесконечность;
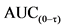 - AUC в интервале дозирования (согласно ОХЛП препарата сравнения);
- AUC в интервале дозирования (согласно ОХЛП препарата сравнения);
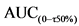 - AUC в ходе первой половины интервала дозирования (согласно СвХП препарата сравнения);
- AUC в ходе первой половины интервала дозирования (согласно СвХП препарата сравнения);
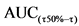 - AUC в ходе второй половины интервала дозирования (согласно ОХЛП препарата сравнения);
- AUC в ходе второй половины интервала дозирования (согласно ОХЛП препарата сравнения);
Cmax - максимальная плазменная концентрация;
tmax - время достижения Cmax;
t1/2 - период полувыведения из плазмы.
Фармакодинамические параметры:
СВГ-AUC(0-t) - площадь под кривой скорости введения глюкозы с момента начала введения и до завершения клэмп-эксперимента в момент t;
СВГ-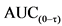 - AUC в интервале дозирования;
- AUC в интервале дозирования;
СВГmax - максимальная скорость введения глюкозы;
tСВГmax - время до достижения максимальной скорости введения глюкозы;
Время до начала действия - время после введения инсулина, когда возникает потребность в первом введении глюкозы с целью поддержания эугликемии, или время после введения инсулина, при котором увеличение СВГ по сравнению с базальной превышает заранее установленный порог отсечения (например, 10 или 20 процентное увеличение СВГ от базальной).
Глава 15.8. Доклинические и клинические исследования
биоаналогичного (биоподобного) лекарственного препарата
рекомбинантного интерферона альфа
1. Введение
В данной главе изложены требования к доклинической и клинической разработке лекарственных препаратов, содержащих рекомбинантный интерферон 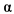 (ИНФ-
(ИНФ- ) и заявленных к регистрации как подобные уже присутствующему на рынке оригинальному (референтному) лекарственному препарату.
) и заявленных к регистрации как подобные уже присутствующему на рынке оригинальному (референтному) лекарственному препарату.
В разделе по доклиническим данным приведены сведения о сравнительной фармакотоксикологической оценке. В разделе по клиническим данным рассматриваются сравнительные исследования фармакокинетики, фармакодинамики, эффективности, безопасности, а также план по управлению рисками.
Молекулы человеческих интерферонов альфа 2a или 2b, состоящие из 165 аминокислот, хорошо изучены и охарактеризованы. Молекулярная масса негликозилированной молекулы равна приблизительно 19 240 Да. Молекула интерферона имеет две дисульфидные связи - одна между остатками цистеина в положении 1 и 98, вторая между остатками цистеина в положении 29 и 138. Первичная структура молекулы содержит потенциальные области O-гликозилирования.
Рекомбинантные интерфероны альфа 2a или 2b (ИНФ- ) рекомендованы для лечения различных заболеваний (например, вирусный гепатит B и C, лейкемия, лимфома, карцинома клеток почечного эпителия и множественная миелома). ИНФ-
) рекомендованы для лечения различных заболеваний (например, вирусный гепатит B и C, лейкемия, лимфома, карцинома клеток почечного эпителия и множественная миелома). ИНФ- применяется для монотерапии и в комбинации с другими препаратами, подтипы 2a и 2b интерферона альфа имеют различные показания для применения. Интерферон альфа может вызывать несколько фармакодинамических эффектов, относительное значение данных эффектов при разных показаниях к применению не ясно. В целом применение интерферона альфа 2a или 2b для лечения онкологических заболеваний значительно снизилось в результате замещения другими методами лечения.
применяется для монотерапии и в комбинации с другими препаратами, подтипы 2a и 2b интерферона альфа имеют различные показания для применения. Интерферон альфа может вызывать несколько фармакодинамических эффектов, относительное значение данных эффектов при разных показаниях к применению не ясно. В целом применение интерферона альфа 2a или 2b для лечения онкологических заболеваний значительно снизилось в результате замещения другими методами лечения.
Для достижения клинического эффекта при применении ИНФ- доза и схема лечения могут значительно различаться в зависимости от заболевания. Обычно ИНФ-
доза и схема лечения могут значительно различаться в зависимости от заболевания. Обычно ИНФ- вводится подкожно, но может вводиться внутримышечно и внутривенно. Лечение интерфероном альфа сопровождается рядом таких нежелательных реакций, как гриппоподобное состояние, усталость и миалгия. Кроме того, ИНФ-
вводится подкожно, но может вводиться внутримышечно и внутривенно. Лечение интерфероном альфа сопровождается рядом таких нежелательных реакций, как гриппоподобное состояние, усталость и миалгия. Кроме того, ИНФ- может вызвать нежелательные реакции, связанные с нарушением психической деятельности, гематологическими и почечными нарушениями.
может вызвать нежелательные реакции, связанные с нарушением психической деятельности, гематологическими и почечными нарушениями.
Терапия интерфероном альфа 2a или 2b может вызвать выработку аутоантител. Применение ИНФ- может привести к развитию таких иммунно-опосредованных состояний, как заболевания щитовидной железы, ревматоидный артрит, системная красная волчанка, невропатии и васкулиты.
может привести к развитию таких иммунно-опосредованных состояний, как заболевания щитовидной железы, ревматоидный артрит, системная красная волчанка, невропатии и васкулиты.
Введение препаратов ИНФ- сопровождается выработкой ненейтрализующих и нейтрализующих антител.
сопровождается выработкой ненейтрализующих и нейтрализующих антител.
2. Сфера применения
Настоящая препарат-специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих рекомбинантный ИНФ- .
.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Доклинические исследования
До начала клинических исследований проводятся доклинические исследования, которые носят сравнительный характер. Основной целью доклинических исследований является выявление возможных различий фармакотоксикологических свойств биоаналогичного (биоподобного) и оригинального (референтного) интерферона альфа, а не простая характеристика данных свойств биоаналогичного (биоподобного) препарата per se. При этом выбор подхода (программа проведения и объем исследований) должен быть полностью обоснован в доклиническом обзоре (модуль 2.4 регистрационного досье).
Исследование фармакодинамики
Исследования in vitro
Исследование фармакодинамики in vitro проводится для сравнительной оценки биологической активности биоаналогичного (биоподобного) и оригинального (референтного) препаратов. Необходимо представить несколько сравнительных биотестов (например, исследования связывания с рецепторами, противовирусные эффекты в культуре клеток, антипролиферативное действие на человеческие опухолевые клеточные линии), многие из этих результатов могут быть получены уже при изучении показателей качества и формировании соответствующего модуля регистрационного досье. Применяемые в подобных исследованиях методы должны быть стандартизированы и валидированы в соответствии с нормативными требованиями.
Необходимо учитывать ограничения при изучении противовирусного действия на клеточных системах, экспрессирующих вирус гепатита C, так как результаты данного теста не коррелируют с клиническим ответом. По возможности для оценки активности и эффективности препаратов необходимо использовать стандартизированные и валидированные методики.
Исследование in vivo
Для подтверждения сопоставимости клинических показаний к применению фармакодинамическая активность биоаналогичного (биоподобного) и оригинального (референтного) препаратов количественно сопоставляется in vivo с помощью следующих методов (методик):
адекватная экспериментальная модель на животных для характеристики фармакодинамических свойств препарата (например, для оценки маркеров фармакодинамических эффектов интерферона, в частности, для определения в сыворотке активности 2',5'-олигоаденилатсинтетазы). Если возможно, данные исследования могут быть проведены в рамках описанных ниже токсикологических исследований;
подходящая модель опухолевого процесса на животных (например, голые ("nude") мыши с ксенотрансплантантной опухолью человека);
подходящая экспериментальная противовирусная модель на животных.
Токсикологические исследования
Изучение токсичности препарата должно включать проведение как минимум одного сравнительного исследования токсичности при многократном введении одной дозы препарата с использованием релевантного вида животных (например, на модели сирийского золотистого хомячка). Продолжительность исследования должна составлять не менее 4 недель.
Исследования токсичности проводятся с учетом требований глав 5.3 и 15.2 настоящих Правил. Следует провести соответствующие измерения токсикокинетики в рамках исследования токсичности при многократном введении препарата, которое должно включать оценку выработки антител (глава 11 настоящих Правил).
Исследования местной переносимости должны быть проведены по крайней мере на одном виде животного. Если имеется возможность, данные исследования проводятся в рамках оценки токсичности при многократном введении одной дозы препарата.
Изучение фармакологической безопасности, влияния на репродуктивность, мутагенности и канцерогенности не входит в перечень стандартных требований к доклиническим исследованиям биоаналогичных (биоподобных) лекарственных препаратов, содержащих человеческий рекомбинантный интерферон альфа.
5. Клинические исследования
Исследование фармакокинетики
Изучение фармакокинетики включает проведение сравнительных перекрестных исследований одной дозы препарата при подкожном и внутривенном введении здоровым добровольцам. Первичным фармакокинетическим параметром, подлежащим оценке является AUC, в качестве вторичных показателей используются Cmax и T1/2 или CL/F. Предельные значения параметров эквивалентности должны быть определены до начала исследования и должным образом обоснованы.
Исследование фармакодинамики
О взаимодействии интерферона альфа с системой иммунитета свидетельствуют следующие маркеры:  -микроглобулин, неоптерин и активность сывороточной 2',5'-олигоаденилатсинтетазы. Сравнительную оценку изучаемых показателей следует проводить для доз, соответствующих возрастающей линейной части кривой "доза - эффект". Так как относительная значимость этих эффектов в отношении разных терапевтических показаний не известна, дополнительная информация может быть получена при комплексной сравнительной оценке этих маркеров после введения биоаналогичного (биоподобного) и оригинального (референтного) препаратов.
-микроглобулин, неоптерин и активность сывороточной 2',5'-олигоаденилатсинтетазы. Сравнительную оценку изучаемых показателей следует проводить для доз, соответствующих возрастающей линейной части кривой "доза - эффект". Так как относительная значимость этих эффектов в отношении разных терапевтических показаний не известна, дополнительная информация может быть получена при комплексной сравнительной оценке этих маркеров после введения биоаналогичного (биоподобного) и оригинального (референтного) препаратов.
Исследование эффективности
Исследуемая целевая популяция
При проведении клинических исследований необходимо учитывать, что интерферон альфа вызывает несколько несвязанных между собой эффектов. Основной целью данного этапа клинического исследования является оценка подобия (сходства) эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов. Это может быть достигнуто при проведении исследований с применением этих препаратов в соответствии с общей характеристикой оригинального (референтного) лекарственного препарата в популяции больных с хроническим гепатитом C (HCV), которые ранее не получали лечения. Другие целевые популяции больных подбираются в зависимости от интересующих показаний, на которые планируется экстраполировать результаты клинических исследований (в соответствии с разделом 6 настоящей главы).
Дизайн и продолжительность исследования
Оценка эффективности проводится в рандомизированном сравнительном исследовании в параллельных группах пациентов, получающих биоаналогичный (биоподобный) и оригинальный (референтный) препараты, продолжительностью как минимум 48 недель. Если позволяют условия, изучение эффективности необходимо проводить в двойном слепом исследовании по крайней мере для получения достаточных данных для проведения первичного анализа. Если использование данного дизайна невозможно, то необходимо в протоколе исследования четко указать обоснование этого и описать мероприятия для снижения (устранения) возможных ошибок статистического заключения (bias).
Режим применения (дозы, путь введения и схема лечения) при проведении клинических испытаний должны быть такими же, как указано в общей характеристике оригинального (референтного) лекарственного препарата. При проведении клинических исследований биоаналогичного (биоподобного) препарата интерферона альфа его применение должно соответствовать рекомендациям, указанным в общей характеристике оригинального (референтного) лекарственного препарата, и текущему стандарту лечения хронического гепатита C.
Программа изучения эффективности препарата должна быть составлена таким образом, чтобы первичная оценка эффективности была проведена на 12-й неделе исследования у всех больных. Предпочтительно проведение исследований в однородной популяции больных, прежде всего по генотипу вируса HCV. Если исследование эффективности проводится в неоднородной популяции больных, то оно должно быть стратифицировано на основе генотипа.
Конечные точки эффективности
Первичная: вирусологический ответ, измеряемый как доля пациентов с уровнем РНК HCV, не определяемым с помощью количественной ПЦР на 12-й неделе. Выбранная методика количественного определения, используемая для измерения уровня РНК HCV, и применяемые пороговые значения также подлежат обоснованию. Сопутствующей конечной точкой может быть снижение вирусной нагрузки в 2 log.
Вторичная: вирусологический ответ на 4-й неделе и в конце лечения, устойчивый вирусологический ответ (через 24 недели после завершения лечения), изменение биохимических показателей печени, включая уровни трансаминаз и заболеваемость.
Исследование безопасности
Сравнительная оценка безопасности проводится в период лечения и в течение 24 недель после его окончания в виде наблюдения за пациентами, получавшими повторные дозы препаратов в ходе сравнительного клинического исследования. Количество испытуемых должно быть достаточным, чтобы можно было провести сравнительную оценку сходства (различия) профиля нежелательных явлений в ответ на введение биоаналогичного (биоподобного) и оригинального (референтного) препаратов. Кроме учета нежелательных явлений изучение безопасности включает оценку изменений лабораторных показателей. Профили безопасности биоаналогичного (биоподобного) и оригинального (референтного) препаратов должны быть подобны в отношении основных нежелательных реакций (например, гриппоподобные симптомы, алопеция, миалгия, лейкопения, анемия и тромбоцитопения).
Иммуногенность
Сравнительная оценка иммуногенности (определение уровня антител) препарата проводится в период лечения и в течение 24 недель после окончания применения препарата в соответствии с требованиями глав 11 и 15.2 настоящих Правил.
При обнаружении антител к препарату необходимо проведение исследований для их характеристики (например, оценка нейтрализующей активности и влияния на эффективность рекомбинантного интерферона альфа). Кроме того, необходимо оценить любую потенциальную возможность нейтрализующего действия антител на активность эндогенных интерферонов (например, развитие аутоиммунных заболеваний). Любое проявление иммуногенности подлежит тщательному изучению в следующих случаях:
при отсутствии ответа на лечение;
при снижении ответа на первичное лечение;
при развитии непредвиденных нежелательных реакций или известных иммуноопосредованных реакций.
6. Экстраполяция результатов исследований
Экстраполяция полученных результатов безопасности и эффективности на другие показания препарата, которые не были изучены в клинических исследованиях, возможна при условии, что механизм действия и (или) рецепторы, участвующие в реализации эффекта для данного показания такие же, как и для показания, по которому сходство в эффективности было установлено в клинических исследованиях.
Если планируется экстраполяция результатов исследования на показание, для которого не доказано сходство механизма действия, то подобная экстраполяция должна быть подробно и полностью обоснована в регистрационном досье.
7. План фармаконадзора
При регистрации препарата необходимо представить в модуль 1 регистрационного досье план управления рисками в соответствии с правилами регистрации и экспертизы лекарственных средств для медицинского применения и правилами надлежащей практики фармаконадзора Союза, утверждаемыми Комиссией.
В пострегистрационном периоде основное внимание должно быть уделено контролю за иммуногенностью, редкими и (или) серьезными нежелательными реакциями в первую очередь среди больных, длительно принимающих препараты. Данные о безопасности должны быть получены на группах пациентов, соответствующих всем одобренным показаниям к применению.
Глава 15.9. Биоаналогичный (биоподобный) лекарственный
препарат интерферона бета
1. Введение
В настоящей главе представлены требования для проведения доклинических и клинических исследований препаратов, содержащих рекомбинантный интерферон бета, для демонстрации подобия (сходства) с уже зарегистрированным оригинальным (референтным) препаратом. В разделе, посвященном проведению доклинических исследований, представлены указания для изучения фармакотоксикологических свойств интерферона бета. В разделе, посвященном клиническим исследованиям, содержатся указания для оценки фармакодинамических и фармакокинетических свойств интерферона бета, эффективности и безопасности и вопросы фармаконадзора.
Рекомбинантный интерферон бета (ИФН- -1a) представляет собой гликозилированную полипептидную цепь, содержащую 166 аминокислотных остатков. Зарегистрированные на сегодняшний день препараты ИФН-
-1a) представляет собой гликозилированную полипептидную цепь, содержащую 166 аминокислотных остатков. Зарегистрированные на сегодняшний день препараты ИФН- -1a различаются между собой по молекулярной структуре, способу введения, терапевтическим дозам и показаниям при рассеянном склерозе.
-1a различаются между собой по молекулярной структуре, способу введения, терапевтическим дозам и показаниям при рассеянном склерозе.
Рекомбинантный интерферон бета ИФН- -1b производится в виде негликозилированной полипептидной цепи, состоящей из 165 аминокислот, и не содержит N-концевого метионина, имеет аминокислотную замену в положении 17 и вводится подкожно.
-1b производится в виде негликозилированной полипептидной цепи, состоящей из 165 аминокислот, и не содержит N-концевого метионина, имеет аминокислотную замену в положении 17 и вводится подкожно.
Лекарственные препараты, содержащие рекомбинантный ИФН- , обычно применяют у пациентов с ремитирующей формой рассеянного склероза, включая лиц с высоким риском развития рассеянного склероза после одного демиелинизирующего события. ИНФ-
, обычно применяют у пациентов с ремитирующей формой рассеянного склероза, включая лиц с высоким риском развития рассеянного склероза после одного демиелинизирующего события. ИНФ- относится к I семейству интерферонов, которые взаимодействуют с рецептором IFNAR, запуская транскрипцию сотен генов.
относится к I семейству интерферонов, которые взаимодействуют с рецептором IFNAR, запуская транскрипцию сотен генов.
Механизм действия ИНФ- при рассеянном склерозе до конца не изучен. Предполагается, что ИНФ-
при рассеянном склерозе до конца не изучен. Предполагается, что ИНФ- действует в качестве иммуномодулятора, нарушая активацию T-клеток по нескольким путям, которые могут включать в себя подавление экспрессии молекул MHC II типа, подавление секреции Th1 лимфоцитами провоспалительных цитокинов, что стимулирует секрецию Th2 лимфоцитами противовоспалительных цитокинов и активирует супрессорные T-клетки, а также препятствуя разрушению гематоэнцефалического барьера и проникновению T-лимфоцитов центральную нервную систему.
действует в качестве иммуномодулятора, нарушая активацию T-клеток по нескольким путям, которые могут включать в себя подавление экспрессии молекул MHC II типа, подавление секреции Th1 лимфоцитами провоспалительных цитокинов, что стимулирует секрецию Th2 лимфоцитами противовоспалительных цитокинов и активирует супрессорные T-клетки, а также препятствуя разрушению гематоэнцефалического барьера и проникновению T-лимфоцитов центральную нервную систему.
Применение рекомбинантного ИФН- при рецидивирующем рассеянном склерозе дает умеренный эффект и снижает частоту обострений по сравнению с плацебо только на 30 процентов, а данные о влиянии препарата на прогрессирование инвалидизации противоречивы.
при рецидивирующем рассеянном склерозе дает умеренный эффект и снижает частоту обострений по сравнению с плацебо только на 30 процентов, а данные о влиянии препарата на прогрессирование инвалидизации противоречивы.
Все препараты ИФН- вызывают схожие нежелательные реакции, которые могут повлиять на приверженность пациента к лечению. Наиболее частыми нежелательными реакциями являются гриппоподобные симптомы: лихорадка, озноб, артралгия, недомогание, потливость, головная боль и миалгия. При подкожном введении препаратов ИФН-
вызывают схожие нежелательные реакции, которые могут повлиять на приверженность пациента к лечению. Наиболее частыми нежелательными реакциями являются гриппоподобные симптомы: лихорадка, озноб, артралгия, недомогание, потливость, головная боль и миалгия. При подкожном введении препаратов ИФН- чаще встречаются реакции в месте введения, бессимптомные нарушения печени и лейкоцитов. Менее распространенными нежелательными реакциями являются депрессия и аутоиммунные нарушения, проявляющиеся нарушением функции щитовидной железы или печени. Все препараты ИФН-
чаще встречаются реакции в месте введения, бессимптомные нарушения печени и лейкоцитов. Менее распространенными нежелательными реакциями являются депрессия и аутоиммунные нарушения, проявляющиеся нарушением функции щитовидной железы или печени. Все препараты ИФН- вызывают выработку антител, особенно нейтрализующих антител. Выработка антител к препаратам варьируется в широких пределах, при проведении клинических исследований было установлено, что при внутримышечном еженедельном введении ИФН-
вызывают выработку антител, особенно нейтрализующих антител. Выработка антител к препаратам варьируется в широких пределах, при проведении клинических исследований было установлено, что при внутримышечном еженедельном введении ИФН- антитела встречаются в 5 процентах случаев, а при подкожном введении ИФН-
антитела встречаются в 5 процентах случаев, а при подкожном введении ИФН-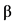 -1b через день - в 45 процентах случаев. В большинстве случаев антитела образуются в течение первого года лечения, но они могут оказывать влияние на эффективность лечения после 18 - 24 месяцев лечения.
-1b через день - в 45 процентах случаев. В большинстве случаев антитела образуются в течение первого года лечения, но они могут оказывать влияние на эффективность лечения после 18 - 24 месяцев лечения.
2. Сфера применения
Настоящая препарат специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) 2-лекарственных препаратов, содержащих ИФН-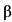 .
.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Основной текст документа
4.1. Доклинические исследования
При планировании доклинических исследований следует применить пошаговый подход для оценки сходства биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов.
Доклинические исследования нужно проводить перед началом клинических исследований. Исследования in vitro должны быть проведены в первую очередь, по их результатам должно быть сделано заключение о том, какие потребуются (если потребуются) исследования in vivo. Принятый подход должен быть полностью обоснован в доклиническом обзоре (модуль 2.4 регистрационного досье).
Исследования in vitro
Для выявления различий в биологической активности биоаналогичного (биоподобного) и оригинального (референтного) препаратов необходимо провести сравнительную оценку специфической активности препаратов в ряде биотестов (например, исследование способности связываться с рецептором, изучение противовирусного, анти пролиферативного и иммуномодулирующего действия препарата). Некоторые из данных показателей могут быть получены в процессе оценки качества препаратов (для препаратов ИФН-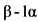 соответствующая статья Фармакопеи Союза).
соответствующая статья Фармакопеи Союза).
Доклинические исследования носят сравнительный характер, должны иметь достаточную чувствительность для выявления различий в ответе между биоаналогичным (биоподобным) и оригинальным (референтным) лекарственными препаратами и не должны лишь оценивать ответ как таковой. Они должны быть проведены на достаточном числе репрезентативных серий препарата, который планируется использовать в клинических исследованиях.
Везде, где возможно, аналитические методы должны быть стандартизированы и валидированы в соответствии с действующими нормативными требованиями (например, оценка противовирусного действия в культуре клеток согласно Фармакопее Союза).
Исследования in vivo
Как правило, проведение экспериментальных исследований на животных не требуется.
Если по результатам оценки качества и (или) выполненных биотестов (фармакологических исследований) in vitro подобие биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов окончательно установить не удается, необходимо проведение дополнительных экспериментальных исследований.
Программа исследования in vivo должна быть разработана специально для оценки выявленных различий и может включать в себя изучение фармакологических свойств и (или) токсичности при многократном введении на релевантных видах животных.
Дальнейшие исследования с использованием релевантных видов животных следует проводить в том случае, если предполагается, что они позволят получить дополнительную информацию.
4.2. Клинические исследования
Клинические сравнительные исследования проводятся поэтапно, начиная с изучения фармакокинетики и фармакодинамики и последующей оценки эффективности и безопасности.
Исследование фармакокинетики
Сравнительное изучение фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов проводится в перекрестном исследовании для каждого пути введения препарата. Оптимальной популяцией для изучения фармакокинетики являются здоровые добровольцы. Доза препарата должна быть подобрана таким образом, чтобы позволить провести сравнение в восходящей линейной области кривой "доза - эффект". Если отсутствует полная информация об оригинальном (референтном) препарате, то предпочтительным является изучение более чем 1 дозы препарата. При оценке фармакокинетических свойств необходимо обоснование выбранного режима введения препарата: однократное или многократное введение (например, 3 раза в неделю). Предпочтительным является использование однократного введения при условии, что методы оценки фармакокинетики достаточно чувствительные, чтобы охарактеризовать полный фармакокинетический профиль. Несмотря на то, что при изучении фармакокинетики после нескольких введений ИФН-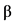 не предполагается выработки антител, до и после проведения каждого курса введения препарата необходимо определение антител, любых возможных расхождений с истинным фармакокинетическим профилем.
не предполагается выработки антител, до и после проведения каждого курса введения препарата необходимо определение антител, любых возможных расхождений с истинным фармакокинетическим профилем.
Концентрация ИФН- после его введения в терапевтических дозах в сыворотке очень низкая и ее измерение является технически сложным. Учитывая это, можно использовать метод определения белка A резистентности к миксовирусам (MxA) на клетках, который позволяет количественно охарактеризовать биологическую активность ИФН-
после его введения в терапевтических дозах в сыворотке очень низкая и ее измерение является технически сложным. Учитывая это, можно использовать метод определения белка A резистентности к миксовирусам (MxA) на клетках, который позволяет количественно охарактеризовать биологическую активность ИФН- в образцах сыворотки крови, и определение ИФН-
в образцах сыворотки крови, и определение ИФН- методом иммуноферментного анализа. При этом необходимо обосновать рациональность выбора метода исследования.
методом иммуноферментного анализа. При этом необходимо обосновать рациональность выбора метода исследования.
Сравнительная оценка фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов проводится на основании показателей AUC, Cmax и T1/2 или Cl/F. Предел эквивалентности должен быть определен заранее и соответствующим образом обоснован, особенно принимая во внимание высокую изменчивость соответствующих фармакокинетических параметров. При планировании в протокол исследования можно включить 2-этапную схему проведения исследования при условии использования откорректированных уровней значимости для каждого из анализов в соответствии с Правилами проведения исследований биоэквивалентности лекарственных препаратов в рамках Союза.
Исследование фармакодинамики
Оптимальной является оценка фармакодинамики в рамках сравнительных исследований фармакокинетики. В настоящее время не установлен биологический маркер, который связан с влиянием ИФН- на течение рассеянного склероза. Однако хорошо описаны маркеры биологической активности ИФН-
на течение рассеянного склероза. Однако хорошо описаны маркеры биологической активности ИФН- , которые можно использовать для всесторонней сравнительной оценки подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов (так называемый подход на основе "отпечатков пальцев"). В настоящее время наиболее чувствительным маркером биологической активности интерферонов I типа является белок A резистентности к миксовирусам (MxA), который можно оценить в крови и как сам белок, и как его мРНК. Кроме данного маркера к показателям, характеризующим фармакодинамические свойства ИФН-
, которые можно использовать для всесторонней сравнительной оценки подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов (так называемый подход на основе "отпечатков пальцев"). В настоящее время наиболее чувствительным маркером биологической активности интерферонов I типа является белок A резистентности к миксовирусам (MxA), который можно оценить в крови и как сам белок, и как его мРНК. Кроме данного маркера к показателям, характеризующим фармакодинамические свойства ИФН- , относится неоптерин, который позволяет оценить зависимости "доза - эффект", также для оценки фармакодинамики ИФН-
, относится неоптерин, который позволяет оценить зависимости "доза - эффект", также для оценки фармакодинамики ИФН- могут быть использованы активность сывороточной 2',5'-олигоаденилатсинтетазы, уровень интерлейкина-10 и TNF-подобного лиганда, индуцирующего апоптоз (TRAIL).
могут быть использованы активность сывороточной 2',5'-олигоаденилатсинтетазы, уровень интерлейкина-10 и TNF-подобного лиганда, индуцирующего апоптоз (TRAIL).
Информативным методом мониторинга очагов демиелинизации при рассеянном склерозе является магнитно-резонансная томография (МРТ). Показатели МРТ связаны с клиническими заболеваниями (например, области T1, накапливающие гадолиний, свидетельствуют о повреждении, а новые или повторяющие области T2 - об увеличении очагов повреждения или рецидивах).
Исследование эффективности
При проведении клинических исследований необходимо продемонстрировать подобную (сходную) эффективность биоаналогичного (биоподобного) и оригинального (референтного) препаратов в рандомизированном параллельном, предпочтительно двойном слепом клиническом исследовании достаточной статистической мощности. Если технически невозможно провести ослепление, необходимо принять дополнительные меры, чтобы избежать получения ошибки в оценке результатов. Способ введения биоаналогичного (биоподобного) препарата должен соответствовать способу, указанному в общей характеристике оригинального (референтного) препарата.
Приемлемый показатель первичной эффективности препарата, модифицирующего течение рассеянного склероза в ремиттирующей форме, число рецидивов заболевания, которое использовалось в базовых клинических исследованиях лекарственных препаратов, содержащих рекомбинантный ИФН- -1a. Несмотря на то, что данный показатель является наиболее предпочтительным для оценки эффективности, он не является необходимым в контексте исследования биоподобия, поскольку это исследование направлено на демонстрацию сопоставимого клинического действия биоаналогичного (биоподобного) и оригинального (референтного) препаратов, которая затем позволит экстраполировать показатель польза-риск оригинального (референтного) препарата. Для демонстрации подобия (сходства) эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов достаточно использовать [магнитно-резонансную томографию] (МРТ) поражений, возникших при рассеянном склерозе в ремиттирующей форме (см. Исследование фармакодинамики). В дополнение к этому такие клинические эффекты, как число рецидивов или процент пациентов без рецидивов, нужно использовать в качестве вторичных конечных точек в поддержку результатов МРТ.
-1a. Несмотря на то, что данный показатель является наиболее предпочтительным для оценки эффективности, он не является необходимым в контексте исследования биоподобия, поскольку это исследование направлено на демонстрацию сопоставимого клинического действия биоаналогичного (биоподобного) и оригинального (референтного) препаратов, которая затем позволит экстраполировать показатель польза-риск оригинального (референтного) препарата. Для демонстрации подобия (сходства) эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов достаточно использовать [магнитно-резонансную томографию] (МРТ) поражений, возникших при рассеянном склерозе в ремиттирующей форме (см. Исследование фармакодинамики). В дополнение к этому такие клинические эффекты, как число рецидивов или процент пациентов без рецидивов, нужно использовать в качестве вторичных конечных точек в поддержку результатов МРТ.
Дизайн исследования эквивалентности должен обеспечить достаточную его чувствительность, т.е. выбор дизайна исследования, участников, длительности и конечных точек МРТ должны сделать возможным выявление различий биоаналогичного (биоподобного) и оригинального (референтного) препаратов, если такие различия фактически существуют. В отношении дизайна исследования чувствительность анализа можно доказать путем проведя исследование с участием 3 групп, включая группу плацебо, в течение короткого периода времени (например, 4 месяца), достаточного для того, чтобы продемонстрировать преимущество как биоаналогичного (биоподобного), так и оригинального (референтного) препаратов над плацебо, используя конечную точку МРТ. В дальнейшем пациенты, которые получали плацебо, должны быть распределены в группы больных, которые получают биоаналогичный (биоподобный) или оригинальный (референтный) препарат.
Альтернативная схема может включать в себя исследование на 3 группах, в котором будут использоваться оригинальный (референтный) препарат (1 группа) и биоаналогичный (биоподобный) препарат в 2 дозировках (2 группы). При этом предполагается, что различия показателей МРТ и клинические исходы будут наблюдаться в течение 12 месяцев. Если при использовании разных доз препарата не будет отмечаться различий в показателях МРТ, то полученные результаты поставят под сомнение чувствительность данного дизайна исследования.
Независимо от программы продолжительность исследования должна быть достаточной, чтобы провести сравнительную оценку эффективности по показателям МРТ и клиническим исходам, т.е. не менее 12 месяцев.
Необходимо подобрать наиболее чувствительную популяцию пациентов, которая позволила бы выявить возможные различия между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами. Это должны быть однородные выборки больных с подтвержденным диагнозом "рецидивирующе-ремиттирующий рассеянный склероз", с активностью процесса по показателям частоты рецидивов и (или) данных МРТ, которые позволят оценить быстрые изменения показателей МРТ.
Изменения показателей МРТ представляют собой приемлемые первичные конечные точки в рамках сравнения биоаналогичного (биоподобного) препарата, если они подкрепляются связанными с рецидивом клиническими исходами. Для полученных клинически значимых результатов не требуется формального испытания эквивалентности; они должны подтвердить тенденцию эффекта аналогичную той, которая наблюдается у изменения показателей, выявленных МРТ. При этом следует четко определить и дифференцировать рецидив от псевдообострения. В процессе исследования необходимо проведение повторных МРТ исследований. При этом необходимо предпринять все меры для обеспечения высокого качества и максимальной надежности измерений. Результаты МРТ должны быть ослепленными и оцениваться (расшифровываться) в одном центре (централизованно). Комбинированные уникальные активные очаги (определяемые как новые T1-взвешенные очаги, накапливающие гадолиний, и новые (увеличивающиеся) T2-взвешенные очаги без их раздельного подсчета) являются наиболее чувствительными критериями эффективности при использовании МРТ и должны рассчитываться при всех исследованиях. При этом может потребоваться кумулятивная оценка ряда изображений (сканограмм). При представлении достаточного обоснования в качестве первичной конечной точки допускается использование других показателей МРТ.
Границы эквивалентности для первичной конечной точки МРТ должны быть заранее установлены и в достаточной мере обоснованы на основании данных МРТ для оригинального (референтного) лекарственного препарата по сравнению с плацебо, или, если они недоступны, на основании экстраполяции других данных по ИНФ- . Следует отметить, что эти данные необходимы на этапе планирования исследования, но не требуются при интерпретации его результатов, поскольку в процессе исследования нужно провести анализ чувствительности. Особое внимание при организации исследования следует уделить критериям выбытия из исследования, потенциальному числу преждевременно выбывающих из исследования пациентов и способу управления отсутствующими данными (методам их обработки).
. Следует отметить, что эти данные необходимы на этапе планирования исследования, но не требуются при интерпретации его результатов, поскольку в процессе исследования нужно провести анализ чувствительности. Особое внимание при организации исследования следует уделить критериям выбытия из исследования, потенциальному числу преждевременно выбывающих из исследования пациентов и способу управления отсутствующими данными (методам их обработки).
Исследование безопасности
Исследование безопасности биоаналогичного (биоподобного) препарата может быть проведено в рамках изучения сравнительной эффективности и является достаточным для исследования более частых нежелательных реакций и формирования базы данных по безопасности перед выпуском препарата на рынок для таких реакций, но не подходит для более редких нежелательных реакций, которые нужно оценить после выпуска препарата на рынок.
Учитывая, что все препараты ИНФ- являются иммуногенными, при проведении клинических исследований проводится оценка иммуногенности препарата на основании главы 11 настоящих Правил. Основная цель такой оценки - сравнение иммуногенности биоаналогичного (биоподобного) и оригинального (референтного) препаратов на протяжении определенного периода времени, так как характеристики антител и последствия их появления могут изменяться с течением времени в результате изменения аффинности и (или) изменения эпитопа. Для регистрации биоаналогичного (биоподобного) препарата необходимо представить данные изучения иммуногенности, проводимого как минимум в течение 12 месяцев. Дальнейшая оценка иммуногенности проводится в пострегистрационном периоде в течение как минимум 6 месяцев. Для оценки сопоставимости динамики образования антител во время лечения необходимо определить стратегию отбора проб, включающую в себя отбор сыворотки в начале исследования и через строго определенные регулярные интервалы (например, каждый месяц в начале лечения (в первые 3 месяца), а затем каждые 3 месяца).
являются иммуногенными, при проведении клинических исследований проводится оценка иммуногенности препарата на основании главы 11 настоящих Правил. Основная цель такой оценки - сравнение иммуногенности биоаналогичного (биоподобного) и оригинального (референтного) препаратов на протяжении определенного периода времени, так как характеристики антител и последствия их появления могут изменяться с течением времени в результате изменения аффинности и (или) изменения эпитопа. Для регистрации биоаналогичного (биоподобного) препарата необходимо представить данные изучения иммуногенности, проводимого как минимум в течение 12 месяцев. Дальнейшая оценка иммуногенности проводится в пострегистрационном периоде в течение как минимум 6 месяцев. Для оценки сопоставимости динамики образования антител во время лечения необходимо определить стратегию отбора проб, включающую в себя отбор сыворотки в начале исследования и через строго определенные регулярные интервалы (например, каждый месяц в начале лечения (в первые 3 месяца), а затем каждые 3 месяца).
Обязательным условием оценки иммуногенности является использование высокочувствительных валидированных методик для выявления и характеристики всех антител (определение класса, подкласса и свойств антител). Необходимо использовать подходы, не допускающие специфической маскировки определенных эпитопов (антигенных детерминант), чтобы предотвратить получение ложноотрицательных результатов. При выявлении антител к ИНФ- в сыворотке больного необходимо оценить нейтрализующую активность и перекрестную реактивность данных антител. Необходимо использовать стандартизированный тест на наличие нейтрализующих антител к белку MxA или метод определения нейтрализующих антител, валидированный относительно этого теста.
в сыворотке больного необходимо оценить нейтрализующую активность и перекрестную реактивность данных антител. Необходимо использовать стандартизированный тест на наличие нейтрализующих антител к белку MxA или метод определения нейтрализующих антител, валидированный относительно этого теста.
Должен быть описан подход, используемый для определения чувствительности анализа (например, при использовании различных пределов), при этом данные определения титров должны быть представлены для каждого временного интервала и каждой исследуемой группы больных. Кроме того, на основании заранее установленных критериев больные должны быть разделены на группы в зависимости от времени развития иммунной реакции. Например, по уровню и свойствам нейтрализующих антител больные могут быть разделены на группы "негативные" (отрицательный ответ у всех образцов после проведения лечения в соответствии с заранее определенными низкими и высокими разведениями или титрами) и "позитивные", которые могут быть разделены на "транзиторно-позитивные" (1 или более образцов после проведения лечения позитивные, сопровождаются негативными образцами на всех последующих или по крайней мере на 2 точках отбора проб) или "постоянно позитивные" (2 или более образцов после лечения постоянно позитивные). Активность на МРТ и клинические рецидивы должны быть сравнимы между этими категориями как для биоаналогичного (биоподобного), так и для оригинального (референтного) препаратов. Влияние нейтрализующих антител на клинические результаты можно установить только по прошествии 12 месяцев терапии, поэтому его необходимо оценивать и после регистрации препарата в рамках плана управления рисками.
Предполагается, что иммунные ответы биоаналогичного (биоподобного) и оригинального (референтного) препаратов будут подобны (сходны) по частоте развития и уровню титров антител (нейтрализующие или ненейтрализующие) и их влияния на эффективность. Несмотря на то, что влияние на эффективность препаратов связывающих (ненейтрализующих) антител до конца неизвестно, повышенная частота появления данных антител при введении биоаналогичного (биоподобного) препарата (относительно оригинального (референтного)) не позволяет рассматривать разрабатываемый препарат как биоаналогичный (биоподобный). Выявление более низкой иммуногенности биоаналогичного (биоподобного) препарата в сравнении с оригинальным (референтным) препаратом необходимо обосновать. При этом препарат может рассматриваться как биоаналогичный (биоподобный) при условии, что его эффективность сопоставима у различных категорий пациентов согласно их иммунному ответу (как ранее определено) и все остальные представленные данные (оценка качества, доклинические исследования, изучение фармакокинетики, фармакодинамики и безопасности) подтверждают биоподобие.
4.3. Фармаконадзор
При регистрации препарата необходимо представить в модуль 1 регистрационного досье план управления рисками в соответствии с Правилами регистрации и экспертизы лекарственных средств для медицинского применения и надлежащей практикой фармаконадзора, в котором должны быть представлены выявленные и потенциальные риски, связанные с использованием оригинального (референтного) лекарственного препарата, и безопасность при утвержденных для него показаниях, основываясь на экстраполяции. План управления рисками должен оценить редкие события (аутоиммунные расстройства, нежелательные реакции и особенно такие, как гепатотоксичность и депрессия, потенциальные эффекты нежелательной иммуногенности и важные отсутствующие данные, например, безопасность при беременности (указание особенностей применения при беременности для препаратов ИНФ- является обязательным)). Этими событиями можно управлять при помощи рутинного фармаконадзора, подкрепленного расширением предрегистрационных исследований (особенно в отношении иммуногенности, как было указано ранее), специальным наблюдательным исследованием или включением в существующий реестр.
является обязательным)). Этими событиями можно управлять при помощи рутинного фармаконадзора, подкрепленного расширением предрегистрационных исследований (особенно в отношении иммуногенности, как было указано ранее), специальным наблюдательным исследованием или включением в существующий реестр.
4.4. Экстраполяция результатов исследований
Экстраполяция результатов исследования клинической эффективности и безопасности, полученных при лечении подтвержденной ремиттирующе-рецессирующей формы рассеянного склероза, биоаналогичного (биоподобного) препарата на другие показания, указанные для оригинального (референтного) препарата, возможна на основе совокупности доказательств, полученных при изучении подобия (сходства).
Глава 15.10. Доклинические и клинические исследования
биоаналогичного (биоподобного) лекарственного препарата
соматотропного гормона
1. Введение
Настоящая глава является дополнением к главе 15.2 настоящих Правил и устанавливает требования проведения доклинических и клинических исследований для демонстрации подобия (сходства) лекарственных препаратов, содержащих соматотропин и заявленных как биоаналогичные (биоподобные) уже представленному на рынке оригинальному (референтному) препарату.
В разделе, посвященном проведению доклинических исследований, представлены рекомендации для оценки фармако-токсикологических свойств препаратов. В разделе, посвященном клиническим исследованиям, содержатся указания для демонстрации подобия (сходства) фармакодинамических, фармакокинетических свойств, эффективности и безопасности 2 препаратов и план управления рисками. Также представлены критерии для экстраполяции клинических данных на другие показания, утвержденные для оригинального (референтного) препарата.
Регистрационное досье нового рекомбинантного гормона роста человека (соматотропина), заявленного в качестве подобного уже представленному на рынке лекарственному препарату, должно содержать доказательства сопоставимости рассматриваемого нового препарата оригинальному (референтному) лекарственному препарату, уже зарегистрированному в Союзе.
Соматотропин продуцируется клетками передней доли гипофиза и представляет собой аминокислотную негликозилированную цепочку, состоящую из 191 аминокислоты, с молекулярной массой 22 кДа. В клинической практике применяется рекомбинантный гормон роста человека (рчГР), который имеет аналогичную с эндогенным аминокислотную последовательность и производится с использованием технологии рекомбинантной ДНК в системе экспрессирующей клеток E. coli, дрожжей или клеток млекопитающих. Для характеристики структуры и биологической активности соматотропина доступны соответствующие физико-химические и биологические методы. Ряд методик и биологических тестов необходимо использовать для характеристики активной фармацевтической субстанции и таких родственных примесей, как дезамидированные и окисленные формы и агрегаты.
Соматотропин обладает мощным анаболическим, липолитическим и контринсулярным (острая инсулиноподобная активность) эффектами. Данные эффекты обусловлены как непосредственным взаимодействием с рецепторами (например, на адипоцитах и гепатоцитах), так и косвенно в результате стимуляции инсулиноподобных факторов роста (преимущественно ИФР-1). Препараты, содержащие в качестве действующего вещества соматотропины, используются в клинической практике для стимуляции нормального роста и (или) формирования тела у больных с дефицитом соматотропина и при некоторых состояниях без дефицита соматотропина. Считается, что при применении рекомбинантного гормона роста человека при всех одобренных в настоящее время показаниях он взаимодействует с одними и теми же рецепторами.
Препараты рчГР используются в широком диапазоне доз для лечения детей в период их роста, в то же время взрослые больные являются более чувствительными к побочным эффектам препарата.
Описаны случаи выработки антител в ответ на введение рчГР, в том числе очень редко - нейтрализующих антител. Это в основном было связано с чистотой и стабильностью применяемых препаратов. Препараты рчГР вводят подкожно, возможные факторы риска развития иммунного ответа, связанные с особенностями состояния больных, не известны.
2. Сфера применения
Настоящая препарат специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) 2 лекарственных препаратов, содержащих рчГР.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Основной текст документа
4.1. Доклинические исследования
До проведения клинических исследований необходимо провести сравнительные доклинические исследования. Основной целью доклинических исследований является выявление возможных различий фармакотоксикологических характеристик биоаналогичного (биоподобного) и оригинального (референтного) препаратов гормона роста, а не изучение результатов как таковых. Выбор подхода к методу исследования должен быть полностью обоснован в доклиническом обзоре (модуль 2.4 регистрационного досье).
Исследование фармакодинамики
Исследование in vitro
Для оценки любых возможных различий биологической активности между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами необходимо представить данные, полученные при проведении ряда сравнительных биотестов (например, исследований связывания с рецепторами, оценки пролиферации клеток), многие из которых могут быть получены в процессе изучения качества препаратов.
Исследование in vivo
Сравнительные исследования in vivo фармакодинамических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов проводятся с использованием экспериментальных моделей (на грызунах). Например, анализируется динамика увеличения массы тела и (или) роста большеберцовой кости у неполовозрелых гипофизэктомированных крыс. Такие данные могут быть получены в процессе выполнения тестов, связанных с оценкой качества препаратов.
Токсикологические исследования
Токсикологические исследования должны включать в себя по крайней мере 1 сравнительное исследование 1 дозы при многократном введении с использованием релевантных видов животных (например, крыс) при длительности исследования не менее 4 недель. В процессе проведения исследований выполняются соответствующие измерения токсикокинетики препарата, особое внимание необходимо уделить изучению иммунного ответа на препарат.
Необходимо представить данные изучения местной переносимости по крайней мере у 1 вида животных. Если это возможно, данные исследования могут быть проведены в процессе оценки токсичности при многократном введении препарата.
Изучение фармакологической безопасности, репродуктивной токсичности, мутагенности и канцерогенности не входит в перечень стандартных требований к доклиническим исследованиям подобных биологических лекарственных препаратов, содержащих в качестве активной фармацевтической субстанции рчГР.
4.2. Клинические исследования
Исследование фармакокинетики
Сравнительная оценка фармакокинетических свойств рчГР проводится в одном перекрестном исследовании биоаналогичного (биоподобного) и оригинального (референтного) препаратов при подкожном введении. В исследование можно привлекать здоровых добровольцев, однако при этом следует рассмотреть необходимость подавления продукции эндогенного гормона роста такими препаратами, как аналоги соматостатина.
К первичным показателям, на основании которых проводится характеристика фармакокинетических свойств препаратов, относится AUC, вторичными показателями фармакокинетики являются Cmax и T1/2. Пределы сопоставимости также должны быть определены заранее и должным образом обоснованы.
Исследование фармакодинамики
Изучение фармакодинамических свойств препарата предпочтительно проводить в рамках исследования фармакокинетики. Для этого необходимо подобрать дозу таким образом, чтобы она соответствовала линейной восходящей части кривой "доза - эффект". Наиболее подходящим маркером для сравнительной оценки фармакодинамических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов является ИФР-1. Кроме того, для оценки фармакодинамики могут быть использованы и другие маркеры, например, белок, связывающий инсулиноподобный фактор роста (ИФРВР-3). Следует учитывать, что вследствие отсутствия четкой связи между уровнем ИФР-1 в сыворотке и индуцируемым им ростом тела ИФР-1 не может использоваться как суррогатный маркер эффективности соматотропина в клинических исследованиях.
Исследование эффективности
Сравнительное изучение эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов должно быть продемонстрировано как минимум в 1 рандомизированном параллельном исследовании достаточной статистической мощности. Для исключения системной ошибки исследование эффективности должно быть двойным слепым. Если это невозможно, необходимо закрыть доступ лицу, отвечающему за измерение параметров клинической эффективности, к данным о распределении испытуемых по группам.
Чувствительность к лечению рчГР выше среди больных с дефицитом гормона роста, чем среди больных, у которых отсутствует дефицит гормона роста. Оптимальной и хорошо изученной моделью оценки эффективности является популяция детей с дефицитом гормона роста, которые ранее не получали подобное лечение, поскольку такой подход обеспечит использование чувствительной и хорошо изученной модели. В исследование необходимо включать детей препубертатного периода для исключения трудности в интерпретации данных, связанной с активным ростом в пубертатный период и ростом, обусловленным эффектом препарата. Это достигается лимитированием возраста (по календарному или костному возрасту) при включении в исследование. Важно, чтобы группы исследования были однородными по исходным характеристикам, поскольку это будет влиять на чувствительность исследования и точность конечных точек.
Первичной конечной точкой исследований эффективности является динамика (изменение) скорости роста или отклонения от динамики стандартной скорости роста по сравнению с исходным уровнем за определенный период времени. В качестве вторичной конечной точки рекомендуется показатель отставания от стандартного роста. Следует делать поправку на факторы, влияние которых на рост при лечении соматотропином уже известно.
При проведении сравнительных исследований необходимо не менее 3 раз проводить измерение роста в каждой временной точке исследования и рассчитывать усредненные данные. При проведении измерений роста рекомендуется применение поверенных измерительных приборов, использование стандартных методик, проведение измерений в одно и то же время суток, предпочтительно одним и тем же исследователем. Это позволяет снизить число ошибок измерения и вариабельность.
Для получения достоверных результатов важно проводить измерение роста до исследования и в процессе исследования с помощью одной и той же стандартизованной методики и с использованием одних и тех же валидированных измерительных приборов.
При кратковременных измерениях роста велика вероятность появления ошибок, связанных со значительной вариабельностью кратковременных периодов роста и сезонными колебаниями, поэтому рекомендованная продолжительность фазы сравнения составляет от 6 до 12 месяцев. Вычисление темпов роста при предварительной обработке данных должно осуществляться с учетом данных, полученные в течение не менее чем 6 месяцев и не более чем 18 месяцев. Диапазон параметров сопоставимости должен быть определен и полностью обоснован заранее, главным образом на основе клинических показателей, обеспечивающих достоверность исследования.
4.3. Исследование безопасности
Данных, полученных при наблюдении за пациентами в ходе исследований эффективности, обычно достаточно для создания адекватной предмаркетинговой базы данных по безопасности. Заявитель должен представить сравнительные показатели по иммуногенности, полученные от пациентов, участвовавших в исследовании эффективности, за 12-месячный период с интервалом забора образцов в 3 месяца. Тестирование нужно проводить с использованием валидированных методов, обладающих достаточной специфичностью и чувствительностью.
Кроме того, клиническое изучение безопасности включает в себя контроль содержания ИФР-1, ИФРВР-3, определение инсулина и глюкозы в крови натощак.
4.4. План фармаконадзора
При регистрации препарата необходимо представить в модуль 1 регистрационного досье план управления рисками в соответствии с Правилами регистрации и экспертизы лекарственных средств для медицинского применения и надлежащей практикой фармаконадзора. При этом необходимо учитывать риски, установленные в процессе разработки препарата, потенциальные риски, связанные с иммуногенностью, а также указать, как эти вопросы будут исследованы при наблюдении в пострегистрационный период.
4.5. Экстраполяция результатов исследований
Результаты исследований эффективности и безопасности, полученные в исследованиях, проведенных в популяции детей с дефицитом гормона роста, могут быть экстраполированы на другие показания, указанные для оригинального (референтного) препарата при условии должного обоснования со стороны заявителя.
Глава 15.11. Доклинические и клинические исследования
биоаналогичного (биоподобного) лекарственного препарата
рекомбинантного фолликулостимулирующего гормона
1. Краткий обзор
В данной главе представлены требования к проведению доклинических и клинических исследований для демонстрации подобия (сходства) 2 рекомбинантных препаратов, содержащих в качестве действующего вещества фолликулостимулирующий гормон человека (рчФСГ).
В разделе, посвященном проведению доклинических исследований, представлены рекомендации для изучения фармако-токсикологических характеристик препаратов. В разделе, посвященном клиническим исследованиям, содержатся указания для демонстрации подобия (сходства) фармакодинамических, фармакокинетических свойств, эффективности и безопасности 2 препаратов, содержащих в качестве действующего вещества фолликулостимулирующий гормон, а также по специфическим мерам управления рисками. Представлены критерии экстраполяции клинических данных на другие показания, утвержденные для оригинального (референтного) лекарственного препарата.
2. Введение
В регистрационное досье на вновь разработанный лекарственный препарат, содержащий в качестве действующего вещества рчФСГ и представляемый для регистрации как биоаналогичный (биоподобный) оригинальному (референтному) лекарственному препарату, зарегистрированному в государствах-членах, должны быть включены материалы, обосновывающие доказательства сходства (подобия) указанных препаратов.
Фолликулостимулирующий гормон (ФСГ) секретируется передней долей гипофиза и играет ключевую роль в регуляции репродуктивных функций женщин и мужчин. ФСГ является гетерогенным димерным гликопротеиновым гормоном, состоящим из 2 связанных между собой субъединиц. Альфа-субъединица, состоящая из 92 аминокислот, подобна субъединицам других гормонов, имеющих гликопротеиновое строение, а бета-субъединица, состоящая из 111 аминокислот, специфична только для ФСГ. Обе субъединицы содержат олигосахаридные структуры. В зависимости от строения и количества углеводных цепочек выделяют несколько изоформ ФСГ с различным содержанием остатков сиаловых кислот. Изоформы с высоким содержанием сиаловых кислот имеют больший период полувыведения. Для описания характеристик белка имеются физико-химические и биологические методы.
Рекомбинантный человеческий фолликулостимулирующий гормон (рчФСГ) используется при нарушении репродуктивных функций у женщин для стимуляции роста и развития фолликулов яичников и у мужчин для стимуляции и поддержания сперматогенеза. Препарат вводится подкожно или внутримышечно.
Самым частым побочным эффектом применения рчФСГ является развитие синдрома гиперстимуляции яичников (OHSS). Это жизнеугрожающее состояние характеризуется развитием тяжелых асцитов, гемоконцентрацией, повышенной коагуляцией, нарушением электролитного баланса и гипертрофией яичников. Большое число стимулированных фолликулов и высокие концентрации эстрадиола, выделяемого созревшими фолликулами, являются факторами риска для развития синдрома гиперстимуляции яичников.
Препараты рчФСГ обладают низкой иммуногенностью, до настоящего времени отсутствуют сообщения о выработке нейтрализующих антител в ответ на введение рчФСГ. При терапевтическом применении препаратов рчФСГ аллергические реакции наблюдались в 0,2 процента случаев и менее чем у 1 пациента на 10000 при введении 2 различных зарегистрированных препаратов рчФСГ. Местные реакции наблюдались более часто (у 3 процентов и у более чем 1 из 10 пациентов, принимавших 2 различных препарата рчФСГ).
3. Сфера применения
Настоящая препарат специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) 2 лекарственных препаратов, содержащих рчФСГ.
4. Связь с другими главами
В главах 15 - 15.2 содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
5. Доклинические исследования
Доклинические исследования должны быть сравнительными и проводиться перед началом клинических исследований. Основной целью доклинических исследований является выявление возможных различий фармакотоксикологических характеристик биоаналогичного (биоподобного) и оригинального (референтного) препаратов, а не оценка реакции на введение препарата как таковой. При этом выбор подхода (программа проведения и объем исследований) должен быть полностью обоснован в доклиническом обзоре (модуль 2.4 регистрационного досье).
Исследования фармакодинамики
Исследование in vitro
Сравнительные исследования in vitro проводятся для выявления возможных различий фармакодинамических свойств по способности связываться с рецептором и вызывать их активацию между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами. Данные исследования могут быть проведены в рамках изучения биологической (специфической) активности при характеристике показателей качества препарата. Для реализации этой цели существует 2 основных подхода. При первом подходе в качестве клетки мишени используются зернистые (гранулезные) клетки или клетки Сертоли. При втором подходе создается постоянно культивируемая клеточная линия (например, на основе CHO), которая имеет стабильные трансфецированные рецепторы к рчФСГ. Преимущество первого подхода заключается в том, что изучение взаимодействия рчФСГ с рецептором осуществляется в условиях, максимально приближенных к естественным, а недостатком данного подхода является ограниченное число клеток тест-системы, что ограничивает количество повторов и использования различных разведений препаратов рчФСГ, которые необходимы для получения достоверных результатов оценки соотношения "концентрация - ответ".
Второй подход позволяет получать в большом количестве материал для исследования, однако зависит от полученной искусственной клеточной конструкции (трансфецированных клеток). Должна быть показана соответствующая чувствительность биотеста, использованного для проверки сопоставимости и выявления потенциальных отличий. Эксперименты должны быть проведены с достаточным числом разведений на каждую кривую для полной характеристики соотношения взаимосвязи "концентрация - ответ".
В исследованиях in vitro необходимо продемонстрировать способность связывания с рецептором, включая кинетику включения - выключения, а также результаты оценки активации рецепторов, т.е. продукции активатора плазминогена (только в классическом исследовании на первичных гранулезных клетках), или накопления внутриклеточного цАМФ. Возможны иные конечные точки (например, активация гена-репортера).
Должны быть представлены в регистрационное досье исследования связывания, включая кинетику включения - выключения, а также результаты оценки активации рецептора, т.е. выработки активатора плазминогена (только в классическом исследовании на первичных гранулезных клетках), или накопления внутриклеточного цАМФ. Заявитель вправе выбрать другие конечные точки (например, активацию гена-репортера). Заявитель должен обосновать выбранный подход в регистрационном досье.
Исследование in vivo
Рекомбинантный фолликулостимулирующий гормон является высоко-гликозилированным белком, и исследования в in vitro не могут в полной мере отразить свойства препарата, которые проявляются в естественных условиях. Таким образом, чтобы квалифицировать любые потенциальные различия между биоаналогичным (биоподобным) рчФСГ и оригинальным (референтным) препаратом, нужно рассмотреть потребность в дополнительных сравнительных исследованиях in vivo.
В настоящее время активность препаратов рчФСГ оценивается методом калибровки по международному стандарту (или внутреннему референтному стандарту, калиброванному относительно международного стандарта, метод Стилмана - Полея). Поскольку таким способом может быть оценена степень активности как биоаналогичного (биоподобного), так и оригинального (референтного) препарата, число различных проводимых анализов может быть уменьшено при помощи разработки исследования, в котором будут одновременно проведены сравнение биоаналогичного (биоподобного) и оригинального (референтного) лекарственных препаратов и их калибровка относительно референтного стандарта. Это позволит уменьшить вариабельность анализа и является более экономичным в отношении расхода реагентов и лабораторных животных. С помощью метода Стилмана - Полея можно установить биологическую активность, но он не позволяет выявить небольшие различия в степени активности между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами. Оценка конечных точек безопасности например, масса тела и местная переносимость) может быть включена в рамки исследований фармакодинамических свойств in vivo.
Необходимо обосновать применение различных методов для сравнения плейотропных эффектов ФСГ в естественном тканевом окружении (например, исследования ex vivo с использованием культуры целых фолликулов или культуры клеток первичной гранулемы). Такой подход в последующем позволит сократить количество требуемых лабораторных животных, избежать вариабельности результатов, полученных при использовании животных, и представляет возможность оценить многие фармакодинамические показатели.
Токсикологические исследования
Проведение специальных исследований токсичности при многократном введении доз не требуется. В отдельных случаях, например, если в препарате используется новое вспомогательное вещество или вещество, которое плохо изучено, необходимо рассмотреть необходимость проведения дополнительных исследований токсичности.
При проведении доклинических исследований биоаналогичных (биоподобных) препаратов рчФСГ исследования фармакологической безопасности и репродуктивной токсичности не требуются. Оценка местной переносимости обычно не требуется. Однако если в состав препарата включены вспомогательные вещества, которые слабо изучены и для которых опыт применения с предполагаемым путем введения очень мал либо отсутствует, необходимо изучение местной переносимости. В данной ситуации оценку местной переносимости можно провести как часть других исследований in vivo.
6. Клинические исследования
Исследование фармакокинетики
Оценка фармакокинетических свойств биоаналогичного (биоподобного) и оригинального (референтного) препаратов проводится в 1 сравнительном перекрестном исследовании при подкожном введении 1 дозы препаратов на здоровых добровольцах женского пола. Для подготовки программы исследований рекомендуется учитывать требования правил проведения исследований биоэквивалентности воспроизведенных лекарственных препаратов Союза. При этом рекомендуется стимулировать подавление секреции эндогенного чФСГ, например, с помощью агониста гонадотропин-высвобождающего гормона или комбинированных пероральных противозачаточных средств. Доза препарата должна быть обоснована с учетом того, что определение различий между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами возможно в линейной части кривой "доза - эффект". Для оценки фармакокинетических свойств препарата необходимо использовать показатели AUC, Cmax, tmax, t1/2 и клиренса. 90 процентный доверительный интервал отношения показателей AUC и Cmax между оригинальным (референтным) и биоаналогичным (биоподобным) препаратами должен составлять от 80 до 125 процентов, за исключением случаев, когда обосновано использование иного интервала. Для представления данных о других параметрах целесообразно использовать методы описательной статистики. Если отсутствует особая необходимость, проведение отдельных фармакологических исследований с внутримышечным введением препаратов не требуется.
Исследование фармакодинамики
Изучение фармакодинамических свойств препарата должно быть проведено в рамках III фазы клинических исследований.
Исследование эффективности
Подобие (сходство) эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов должно быть продемонстрировано в рандомизированном клиническом исследовании достаточной статистической мощности в параллельных группах. Оптимальной моделью для сравнительного изучения эффективности биоаналогичного (биоподобного) и оригинального (референтного) препаратов является стимуляция мультифолликулярного развития у пациенток, у которых ранее наблюдалась гиперовуляция в рамках таких вспомогательных репродуктивных технологий, как экстракорпоральное оплодотворение (ЭКО), внутрифаллопиевый перенос гамет (GIFT) или внутрифаллопиевый перенос зиготы (ZIFT). Для сравнения эффективности необходимо использовать первый цикл лечения.
Рекомендуется проведение двойных слепых исследований. Если условия не позволяют проведение двойных слепых исследований, необходимо использовать ослепление на этапе оценки результатов. В первую очередь это касается результатов, на которые большое влияние оказывает субъективный фактор, например, ультразвуковые исследования и оценка состояния яйцеклетки и (или) эмбриона. Доза рчФСГ препарата должна быть фиксированной в течение первых 5 дней стимуляции. Допускается использование в протоколе агонистов или антагонистов гонадотропин-высвобождающего гормона.
В качестве первичной конечной точки рекомендуется использовать показатель количества зрелых яйцеклеток (ооцитов). Должна быть продемонстрирована эквивалентная эффективность исследуемого и стандартного препаратов и проспективно определены и подтверждены пределы эквивалентности. Диапазон эквивалентности должен быть определен проспективно. При этом необходимо учитывать, что и чрезмерная, и недостаточная стимуляция могут привести к задержке цикла и отсутствию зрелых яйцеклеток (нулевых ооцитов), которые являются основным показателем первичной конечной точки. Поэтому данные должны быть представлены таким образом, чтобы можно было провести детальное сравнение причин отмены циклов вспомогательных репродуктивных технологий.
В качестве альтернативного подхода для сравнительной оценки эффективности с использованием критерия не меньшей активности, может быть использован показатель нормального течения беременности в течение 10 недель после пересадки эмбриона, который может быть использован и в качестве первичной конечной точки. В данном случае показатель количества зрелых яйцеклеток (ооцитов) должен быть включен в качестве дополнительной первичной конечной точки с соответствующим диапазоном эквивалентности или в качестве наиболее важной вторичной конечной точки.
При определении вторичных конечных точек необходимо учесть следующие особенности:
если количество зрелых яйцеклеток (ооцитов) используется в качестве первичной конечной точки, то показатель нормального течения беременности в течение 10 недель после пересадки эмбриона должен рассматриваться в качестве вторичной конечной точки;
при искусственном оплодотворении доза рчФСГ подбирается на основании данных реакции яичников, что может скрывать различия, связанные с препаратами. Необходимо учитывать коррекцию дозы и возможные различия между дозами биоаналогичного (биоподобного) и оригинального (референтного) препаратов. При анализе вторичных конечных точек необходимо учитывать суммарную дозу препарата рчФСГ, количество дней стимуляции препаратом рчФСГ и процент пациентов, которым требовалось увеличение и уменьшение дозы препарата. Если основные различия между биоаналогичным (биоподобным) и оригинальным (референтным) препаратами касаются применяемой дозировки, то такие препараты не соответствуют концепции биоподобия;
следует изучать вспомогательные параметры, которые используются для демонстрации подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов. Соответствующие конечные точки должны включать в себя количество фолликулов и их распределение по размерам во время лечения и в день индукции овуляции. Следующей конечной точкой, которая позволяет оценить фармакодинамические свойства препарата рчФСГ, является определение количества фолликулов после 5 дней стимуляции препаратом рчФСГ (перед проведением корректировки дозы). Кроме того, в сыворотке необходимо определить уровни ингибина-B, эстрадиола, лютеинизирующего гормона и прогестерона;
необходимо учитывать показатели, характеризующие качественные свойства зрелых яйцеклеток (эмбрионов), при этом необходимо документально фиксировать количество ооцитов (эмбрионов) хорошего качества.
Исследование безопасности
Изучения безопасности биоаналогичного (биоподобного) препарата в процессе исследования эффективности, как правило, бывает достаточно для характеристики профиля нежелательных реакций биоаналогичного (биоподобного) препарата. В первую очередь необходимо обратить внимание на синдром гиперстимуляции яичников и при развитии OHSS подробно описать все его проявления (например, степень тяжести - легкая, средняя и тяжелая) и дифференцировать синдром гиперстимуляции яичников с поздним и ранним началом.
Вероятность развития иммунной реакции на препарат чаще связана с подкожным путем введения и прерывистым (периодическим) введением препарата, чем с однократным внутривенным введением. Оба этих фактора могут применяться к рчФСГ, так как женщины могут получать более одного цикла вспомогательных репродуктивных технологий. Данные по иммуногенности должны представляться по всем женщинам, участвующим в исследовании эффективности, а также по всем женщинам, получающим более одного цикла вспомогательной репродуктивной терапии.
Определение иммуногенности нужно проводить до 3 месяцев после проведения терапии рчФСГ с применением валидированных тестов на основе антител достаточной чувствительности и специфичности. При обнаружении потенциального влияния антител к ФСГ на эффективность и (или) безопасность оно должно быть оценено и должна быть рассмотрена необходимость дальнейшей характеристики этих антител, например, в отношении их нейтрализующего потенциала.
7. Фармаконадзор
При регистрации препарата необходимо представить в модуль 1 регистрационного досье план управления рисками в соответствии с Правилами регистрации и экспертизы лекарственных средств для медицинского применения и надлежащей практикой фармаконадзора. В плане управления рисками биоаналогичного (биоподобного) препарата должны быть учтены выявленные и потенциальные риски, связанные с применением оригинального (референтного) препарата, и, если возможно, безопасность при показаниях, одобренных для оригинального (референтного) препарата, основываясь на экстраполяции данных. При этом план должен содержать детальное обсуждение, каким образом указанные проблемы будут устранены в пострегистрационном периоде.
8. Экстраполяция результатов исследований
Демонстрация эффективности и безопасности исследуемого препарата в сравнении с оригинальным (референтным) препаратом при стимуляции мультифолликулярного развития во время проведения вспомогательной репродуктивной терапии позволяет экстраполировать результаты исследования на другие показания, утвержденные для оригинального (референтного) препарата.
Глава 16. Адъюванты вакцин для лечения
и профилактики заболеваний человека
1. Введение
Адъюванты (иммуностимуляторы или иммуномодуляторы) десятилетиями использовались для усиления иммунного ответа на антигены вакцин. Адъюванты включаются в состав вакцины с целью более быстрого формирования выраженного и длительно сохраняющегося специфического иммунного ответа. Целью включения адъювантов в состав вакцины является усиление, ускорение и продление желательного специфического иммунного ответа на антигены вакцины. Преимущества использования адъювантов в вакцинах заключаются в повышении иммуногенности антигенов, изменении характера иммунного ответа, снижении количества антигена, необходимого для успешной иммунизации, сокращения частоты необходимых повторных иммунизаций и улучшения иммунного ответа у пожилых и ослабленных вакцинированных субъектов. Адъюванты можно выборочно использовать для оптимизации требуемого иммунного ответа, например, по отношению к классам иммуноглобулинов и индукции реакций на основе цитотоксических лимфоцитов или T-хелперов. Кроме того, некоторые адъюванты можно использовать для стимуляции гуморального ответа на поверхности слизистых.
Интерес к адъювантам вакцин объясняется несколькими причинами. Производители вакцин и органы здравоохранения (например, ВОЗ) направляют усилия на улучшение существующих вакцин и разработку новых. В последние годы наметились перспективы появления новых потенциальных вакцин против инфекционных, аллергических и аутоиммунных заболеваний, а также для лечения рака и бесплодия. Во многих случаях из-за низкой иммуногенности вакцин требуются адъюванты. Новые технологии в области аналитической биохимии, возможности очистки на макромолекулярном уровне, использование рекомбинантных технологий и достижения в области глубокого понимания иммунологических механизмов и патогенеза заболеваний помогут улучшить техническую базу для разработки и применения адъювантов для новых вакцин.
Адъюванты могут быть классифицированы по происхождению (натуральные, синтетические или эндогенные), по механизму действия, а также по физическим или химическим свойствам. Классы адъювантов, наиболее часто описываемые в настоящее время, указаны в подразделе 2.2 настоящей главы.
Активность адъювантов является результатом нескольких факторов, и усиленный иммунный ответ, полученный с помощью 1 антигена, как правило, не может быть распространен на другой антиген. Поскольку различные антигены различаются по физическим, биологическим и иммуногенным свойствам, им необходимы соответствующие адъюванты. Выбор адъювантов должен быть обусловлен типом желаемого иммунного ответа и соединен с антигеном таким способом, чтобы обеспечить развитие оптимального типа ответа с минимальными побочными эффектами.
Основное свойство большинства адъювантов заключается в их способности депонировать антиген, то есть адсорбировать его на своей поверхности и длительное время сохранять в организме, защищать антиген от разрушения и элиминации, что увеличивает продолжительность влияния антигена на иммунную систему. Основные аспекты воздействия адъювантов представляют собой способность антигена, определяемую физическими свойствами антигена в вакцине, захват антигена (адъюванта), распределение (направленное воздействие на конкретные клетки), стимуляцию (модуляцию) иммунной системы, которая включает в себя регуляцию как количественных, так и качественных аспектов последующих иммунных реакций, защиту антигена от разложения и выведения.
Наиболее сильные адъюванты содержат в своем составе микроорганизмы ослабленных штаммов или какие-либо субстанции, извлеченные из них. Эти компоненты являются стимуляторами компонентов системы врожденного иммунитета, включая макрофаги и другие антигенпрезентирующие клетки.
Для направленной доставки антигена в лимфоидные органы используют липосомы (липидные пузырьки). Это позволяет точно дозировать антиген и избежать его влияния на структуры, не вовлеченные в формирование иммунного ответа.
Основные механизмы проявления активности адъювантов:
оптимизация презентации антигена, который определяет специфичность действия вакцины;
формирование депо антигена, увеличивающего срок его воздействия на систему иммунитета;
распределение антигена в организме (направленная доставка антигена к клеткам иммунной системы);
прямое стимулирующее (модулирующее) действие на клетки иммунной системы, которые в конечном итоге ответственны за количественные и качественные аспекты стимуляции иммунного ответа;
защита антигена от деградации и элиминации.
В целом механизм действия адсорбентов и мелкодисперсных адъювантов включает в себя презентацию антигена иммунной системе, в то время как микробные, синтетические и эндогенные адъюванты действуют путем прямой стимуляции или модуляции иммунной системы. Помимо их роли в презентации антигена иммунной системе, механизм действия эмульсий заключается в стимуляции медленного высвобождения антигена и его защиты от быстрого выведения. Использование адъювантов продленного действия, например, минеральных солей, сопровождается образованием воспалительного очага в области инъекции, что может привести к синтезу провоспалительных цитокинов и стимуляции врожденного иммунитета, важного для первых этапов иммунного ответа.
Таким образом, оценка качества рецептуры вакцины с адъювантом охватывает такие аспекты, как демонстрация совместимости адъюванта с антигенным компонентом, присутствующим в вакцине, доказательство правильной и стабильной ассоциации антигена с адъювантом, демонстрация отсутствия значительной диссоциации на протяжении срока хранения, степень ассоциации на протяжении срока хранения, влияние адъюванта на анализ компонентов, биохимическую чистоту и пирогенность. В качестве примера ассоциации - адсорбция характерна для гелей гидроксида алюминия, гелей фосфата алюминия, гелей фосфата кальция и ISCOMS, в то время как ионные взаимодействия характерны для заряженных мицелл диметилдиоктадециламмония (DDA). Механизмом ассоциации для эмульсий и липосом является инкапсуляция. Для производных сапонинов или других экстрактов взаимодействие с антигенами является липофильным (гидрофильным) или ионным.
В прошлом разрабатывалось много адъювантов, однако они никогда не использовались для плановой вакцинации из соображений безопасности, например, острой токсичности и возможности отсроченных побочных эффектов. Таким образом, следует сопоставлять преимущества адъювантной вакцины и риск вызываемой ей нежелательной реакции. Нынешняя точка зрения на соотношение риск-польза для вакцинации отдает предпочтение безопасности перед эффективностью, если вакцина предназначена для здорового населения. Однако в группах высокого риска, включая пациентов с онкологическими заболеваниями и СПИД, и для других терапевтических вакцин при существенной пользе от вакцины может быть приемлемым повышенный уровень токсичности. Таким образом, в соответствующих случаях следует предусмотреть доклиническую оценку безопасности.
Даже при отсутствии серьезных нежелательных эффектов в ходе обширных доклинических исследований токсикологических аспектов и безопасности нельзя гарантировать, что новая рецептура вакцины с адъювантом не представляет риска для вакцинированных и не приведет к неожиданным явлениям. Непредсказуемость влияния адъюванта на организм человека является результатом сложного взаимодействия между такими факторами, как путь введения, доза антигена и природа антигена. По этой причине окончательная оценка безопасности новой разработанной рецептуры вакцины может проводиться только на основании клинических исследований.
2. Область применения
В методических рекомендациях отражены вопросы оценки качества, доклинических и клинических исследований новых или уже используемых адъювантов в составе вакцин. Требования к используемым в настоящее время адъювантам (например, гидроксиду алюминия, алюминию алюминия или кальция фосфату), изложенные в данном документе, зависят от особенностей индивидуальных вакцин и должны рассматриваться отдельно в каждом конкретном случае.
2.1. Вакцины
Вакцины, рассмотренные в данном документе, обеспечивают формирование иммунитета против инфекционных заболеваний.
Вакцины могут содержать 1 или более из следующих компонентов:
организмы, инактивированные химическим или физическим способом, сохраняющие адекватные иммуногенные свойства;
живые организмы, которые естественно авирулентны или которые были подвергнуты воздействию для аттенуации их вирулентности, сохраняющие адекватные иммуногенные свойства;
антигены, выделенные из инфекционного агента или секретируемые им;
антигены, полученные методом рекомбинантной ДНК-технологии;
живые рекомбинантные векторы, продуцирующие антигены in vivo при вакцинации хозяина (носителя);
плазмидные ДНК;
антигены, полученные путем химического синтеза in vitro.
Антигены могут находиться в нативном состоянии, могут быть укорочены или модифицированы вследствие мутаций, могут быть обезврежены за счет химических или физических средств и (или) агрегированы, полимеризованы или конъюгированы с носителем (см. также соответствующую статью Фармакопеи Союза). В настоящее время адъюванты не используются в живых вакцинах для медицинского применения, однако этого нельзя исключить в будущем.
Основные положения данных рекомендаций применимы к оценке качества и доклиническим исследованиям по изучению терапевтических вакцин (например, таких антиидиотипических вакцин, как моноклональные антитела, используемые в качестве иммуногенов, противоопухолевых вакцин, аллергенов для специфической иммунотерапии (аллергоидов) и вакцин, используемых для лечения инфицированных лиц), однако клинические аспекты терапевтических вакцин не входят в рамки данного документа.
2.2. Адъюванты
Адъювант вакцины представляет собой компонент, стимулирующий иммунный ответ на антиген и (или) модулирующий желательные иммунные реакции.
Адъюванты включают в себя (но не ограничиваются):
минеральные соли, например, гидроксид алюминия и гель фосфата алюминия или кальция;
масляные эмульсии и составы на основе сурфактантов, например, MF59 (микропсевдоожиженный детергент, стабилизированный эмульсией "масло в воде"), QS21 (очищенный сапонин), AS02 [SBAS2] (эмульсия "масло в воде" + MPL + QS21), Montanide ISA-51 и ISA-720 (стабилизированная эмульсия "масло в воде");
адъюванты в виде твердых частиц, например, виросомы (униламеллярные липосомальные частицы с инкорпорированным гемагглютинином гриппа), AS04 ([SBAS4] соль алюминия с MPL), ISCOMS (структурированный комплекс сапонинов и липидов), полилактид-ко-гликолид (PLG);
микробные дериваты (натуральные или синтетические), например, монофосфорил липида A (MPL), Detox (MPL + структуры клеточной стенки M. Phlei), AGP [RC-529] (синтетический ацилированный моносахарид), DC_Chol (липоидный иммуностимулятор, способный самоорганизовываться в липосомы), OM-174 (производное липида A), CpG-мотивы (синтетические олигонуклеотиды, содержащие иммуностимулирующие CpG-последовательности), модифицированные LT и CT (генетически модифицированные бактериальные токсины для обеспечения нетоксигенного адъювантного эффекта);
эндогенные иммуномодуляторы человека, например, гранулоцитарно-макрофагальный колониестимулирующий фактор (hGM-CSF) или интерлейкин-12 (hIL-12) (цитокины, которые применяются как в форме протеина, так и в виде кодирующих их плазмид), Immudaptin (C3d-тандемная область);
такие инертные частицы, как частицы золота.
В случае разработки новых видов адъювантов на них также будут распространяться требования настоящей Главы.
Активный ингредиент комбинированной вакцины, оказывающий адъювантное действие на другие активные ингредиенты вакцины, не входит в сферу применения данной настоящей главы. Кроме того, в сферу применения данной настоящей главы не входят носители гаптенов, антигены (например, CRM197, менингококковый OMP, столбнячный и дифтерийный анатоксины, которые используются для конъюгирования полисахаридов) и вспомогательные вещества, например, HSA.
В готовой вакцине могут присутствовать несколько адъювантов. Они могут быть объединены с одним из антигенов или всеми антигенами, представленными в вакцине, либо каждый адъювант может быть объединен с одним конкретным антигеном.
В любом случае указания, содержащиеся в настоящей главе, при необходимости применимы к каждому адъюванту и каждой комбинации антиген-адъювант.
3. Качество
Происхождение и природа адъювантов, используемых или разрабатываемых в настоящее время, весьма разнообразны. Например, адъюванты на основе алюминия состоят из простых неорганических соединений, PLG представляет собой полимерный углевод, вирус-липосомные вакцины можно получить из неоднородных вирусных частиц, MDP получают из бактериальной клеточной стенки, сапонины имеют растительное происхождение, сквален получают из печени акулы, а рекомбинантные эндогенные иммуномодуляторы получают из рекомбинантных бактерий, дрожжей или клеток млекопитающих. Следовательно, в данной методической рекомендации нецелесообразно приводить полный список отдельных анализов, которые следует выполнять для любого конкретного адъюванта или комбинации адъювант - антиген. Указания, приведенные ниже, должны использоваться производителями адъювантов (вакцин) в индивидуальном порядке исходя из того, насколько они подходят для их адъюванта. Следует придерживаться соответствующих статей Фармакопеи Союза. Для производителей рекомбинантных белков-адъювантов следует использовать соответствующие требования настоящих Правил, например, по клеточным субстратам (глава 1), вирусологической безопасности (главы 2 - 3), рекомбинантным ДНК и белкам (глава 5).
3.1. Адъюванты
3.1.1. Описание.
Следует подробно описать природу или химический состав адъюванта в регистрационном досье. При использовании более чем 1 адъюванта или в случае если адъювант состоит из более чем 1 компонента следует описать функции каждого адъюванта или компонента в той степени, в какой они известны.
3.1.2. Производство.
Производство адъюванта должно быть описано максимально подробно. Особое внимание должно быть уделено источнику получения адъюванта, особенно если он имеет биологическое происхождение или есть какие-либо особенности его применения.
Следует определить параметры, важные для правильных физических, биохимических, биологических или адсорбционных свойств адъюванта. Особое внимание должно быть обращено на использование любых биологических продуктов, получаемых от крупного рогатого скота (бычья сыворотка и др.), при использовании таких материалов требуется соблюдение требований соответствующих рекомендаций Союза и статей Фармакопеи Союза по минимизации риска передачи возбудителей губчатой энцефалопатии животных через лекарственные средства медицинского и ветеринарного назначения.
3.1.3. Характеристика
Следует описать результаты оценки ряда параметров, используемых для изучения характеристик адъювантов. Необходимо выявить и описать основные параметры. Такие параметры могут являться частью регулярных анализов партий адъюванта.
Кроме того, для изучения характеристик адъюванта следует анализировать другие параметры, и некоторые из них могут также входить в состав регулярных анализов. Параметры, которые определяют адъювант, зависят от природы адъюванта и могут включать в себя в числе прочего:
химический состав (качественный и количественный);
физические характеристики (например, визуальную оценку, плотность, вязкость, pH, размерность и распределение частиц по размерам, поверхностный заряд);
биохимические характеристики;
чистоту (например, содержание эндотоксинов, микробиологическую чистоту, остаточное содержание производственных примесей).
3.1.4. Рутинные исследования.
Перечень показателей качества должен быть обоснован результатами их оценки при характеристике адъюванта, как это указано выше. Должна быть установлена спецификация и определены требования к оцениваемым показателям.
3.1.5. Стабильность.
Оценка соответствующих физико-химических и (или) биологических свойств, основанных на характеристике адъюванта, должна проводиться при изучении стабильности адъюванта во время хранения.
Важными параметрами могут быть степень диссоциации антигена и адъюванта и целостность антигена.
3.2. Комплекс адъювант - антиген
3.2.1. Изучение и производство комплексного препарата.
Объединение антигена с адъювантом является главным ключевым моментом в получении конечного комплекса адъювант - антиген. Механизм и эффективность ассоциации (связывания) между антигеном и адъювантом должны быть определены и описаны. Аспекты, которые важны для биологических свойств комплекса адъювант - антиген (например, адсорбция, характеристики связывания), должны быть идентифицированы и проверены. Если используется более 1 адъюванта при объединении их в комплекс, то должна быть представлена информация о каждом адъюванте и проведены исследования по оценке совместимости вводимых в комплекс адъюванта и антигена.
Если конечная вакцина содержит более 1 комплекса адъювант - антиген, требуется выполнение анализов, соответствующих природе адъювантов (идентичных или нет), в том числе анализа нежелательных эффектов между различными комплексами адъювант - антиген.
Процесс производства комплекса адъювант - антиген должен быть детально описан.
Промежуточный продукт может быть получен в процессе формирования комплекса адъюванта и антигена до сведения компонентов. В другом случае сведение компонентов происходит одновременно на этапе объединения адъюванта и антигена (конечный балк). Альтернативный процесс включает одновременное объединение адъюванта и антигена и розлив готового продукта.
Наполнитель или растворитель, добавленные к комплексу адъювант-антиген во время приготовления конечного балка (формулирования), не должны оказывать негативного влияния на активность вакцины или ассоциацию (связывание) антигена с адъювантом.
В каждом конкретном случае характеристика, рутинный контроль и изучение стабильности промежуточного продукта, конечного балка и готового продукта (когда приемлемо) должны быть выполнены в соответствии с пунктом 3.2.2. настоящих Правил. Изготовитель вакцин должен ясно определить и обосновать испытания, которые выполняются на каждой стадии.
3.2.2. Характеристика.
Комплекс адъювант - антиген должен быть соответствующим образом охарактеризован. Характеристика комплекса может включать в себя соотношение и последовательность объединения антигена с адъювантом, целостность антигена в ассоциации с адъювантом, влияние адъюванта на возможность определения антигена и степень возможности диссоциации комплекса антигена с адъювантом (стабильность). Другие параметры могут включать в себя химические и физические характеристики (например, размер частиц, вязкость).
3.2.3. Обычные (рутинные) исследования.
Тесты для рутинной оценки комплекса адъювант - антиген должны быть определены (идентифицированы), описаны и валидированы. Такие тесты должны быть основаны на параметрах, полученных во время полной характеристики комплекса адъювант - антиген.
3.2.4. Стабильность.
Долгосрочная стабильность комплекса адъювант - антиген должна быть исследована с оценкой соответствующих физических и биохимических свойств. Важными параметрами являются степень диссоциации комплекса антиген - адъювант и его целостность.
3.2.5. Несколько антигенов в комбинации с адъювантом.
Если в готовый препарат вакцины включают антиген в дополнение к антигену, существующему в комбинированной вакцине с адъювантом, необходимо изучить эффект, оказываемый адъювантом на этот дополнительный антиген, используя соответствующие для этого антигена исследования. Точно так же любые эффекты дополнительного антигена следует оценить в отношении его воздействия на комплекс адъювант - антиген.
Если в состав конечного продукта вакцины включают более 1 комплекса антиген - адъювант, исследования проводят в соответствии с происхождением адъюванта (является ли он идентичным тому, который присутствует в вакцине) для того, чтобы исключить побочные эффекты, обусловленные взаимодействием между различными комплексами адъювант - антиген.
3.3. Конечный продукт
Готовый препарат вакцины должен исследоваться по таким показателям, как активность, подлинность и стабильность. Данные исследования должны проводиться в соответствии с требованиями Фармакопеи Союза, а также настоящей главы. Методики для тестирования препарата и изучения стабильности должны быть определены и валидированы.
Взаимодействие адъюванта с антигеном может влиять на результаты некоторых стандартных тестов в случае их использования для исследования на стадии готового продукта или сформулированного конечного балка.
Альтернативные методы исследования должны быть разработаны отдельно, они будут отличаться от методов исследования, проводимых на более ранних стадиях, когда воздействие адъювантов на антиген отсутствует.
4. Доклинические исследования
4.1. Свойства адъюванта
Механизм проявления действия адъювантов:
физическое воздействие на механизмы представления вакцинного антигена;
оптимизация проявлений антигенной активности;
доставка антигена в клетки, отвечающие за инициацию иммунного ответа и координирующие взаимодействие важных компонентов иммунной системы, - антигенпрезентирующие клетки;
воздействие на специфические антигенпрезентирующие клетки (дендритные клетки, клетки Лангерганса, макрофаги и др.), которое проявляется стимулированием Toll-подобных рецепторов за счет аналогов липида A или за счет усиливающего иммунный ответ соединения олигодеоксинуклеотида (ODNs) CpG;
усиление или модулирование иммунного ответа, обусловленное, например, внутриклеточным транспортированием и процессированием антигена, ассоциацией с молекулами MHC класса I или II, экспансией T-лимфоцитов, с продукцией различных цитокинов.
Должно быть дано объяснение механизма действия адъювантов. Повышение интенсивности иммунного ответа на комплекс адъювант-антигена должно быть показано в исследованиях на соответствующей модели животных. Следует установить, воздействует ли адъювант на клетки системы врожденного иммунитета. Кроме того, необходимо выяснить степень активации гуморального и клеточного звена иммунного ответа при использовании адъюванта вместе с антигеном. Данные, полученные при использовании комплекса адъюванта с другими антигенами, могут служить основой для выяснения механизма действия адъюванта. Идеально подходящая модель животных должна продемонстрировать защиту иммунизированных животных при введении смертельной дозы патогенных микроорганизмов (при инфекционной патологии). Если такая модель отсутствует, следует использовать в экспериментах тот вид животных, у которых индуцированный иммунный ответ будет подобен ожидаемому иммунному ответу у человека. В качестве уточняющей информации могут быть использованы сведения из научной литературы.
4.2. Фармакокинетика
Фармакокинетические исследования (т.е. исследования с определением концентрации антигена в сыворотке крови) не требуются. В некоторых случаях, фармакокинетические исследования могут иметь значение для понимания механизма действия адъюванта.
4.3. Изучение токсичности адъювантов
Методология, используемая для изучения токсичности адъювантов, должна соответствовать методологии изучения вакцины. Вакцины с адъювантами могут назначаться при использовании в медицинской практике с интервалом от нескольких недель до нескольких лет. Обычно адъюванты вводятся в небольшом количестве несколько раз за весь период жизни.
Адъювант исследуется в соответствии с его использованием в составе вакцины, и стратегия тестирования должна отражать это использование.
Адъюванты могут использоваться с различными антигенами, часто они не обладают видовой специфичностью.
Адъюванты могут быть связаны с различными антигенами и должны быть изучены на 2 видах животных, если нет других указаний (на грызунах и не грызунах).
Адъюванты, принадлежащие к различным биологическим классам, могут проявлять высокий уровень видовой специфичности (например, некоторые цитокины), поэтому исследовать их более чем на 1 виде животных не имеет смысла. Однако другие адъюванты (например, масляные эмульсии) проявляют меньшую специфичность и принципы токсикологических исследований базируются на общих требованиях для изучения токсичности, т.е. исследования проводят как минимум на 2 видах животных. Результаты, полученные при исследовании на 1 виде животных, будут подтверждены результатами исследования на другом виде животных.
Выбор вида животных зависит прежде всего от выбора антигена, для которого предназначен адъювант. Идеальным является вид животных, на котором уже проводили исследования.
4.3.1. Местная переносимость.
Местнораздражающее действие адъюванта должно быть изучено в зависимости от пути его введения. Например:
для орального и внутриносового пути введения должна быть изучена местная и общая толерантность;
для инъекционных вакцин особое внимание должно быть уделено возможности индукции поздней гранулематозной реакции, например, при использовании частиц или минерального масла.
4.3.2. Индукция гиперчувствительности и анафилактической реакции.
Адъюванты могут проявлять иммуногенные свойства, поэтому следует оценивать возможность развития гиперчувствительности на соответствующих моделях (например, в тестах пассивной кожной анафилаксии [PCA] и активной системной анафилаксии [ASA]). Адъюванты могут индуцировать повышение уровня специфического иммуноглобулина класса E (IgE) к антигену вакцины, поэтому следует оценить возможность индукции реакций гиперчувствительности и анафилаксии на антиген при использовании адъюванта.
4.3.3. Пирогенность.
Адъюванты должны быть проверены относительно их возможного пирогенного эффекта. Альтернативные тесты in vitro для оценки пирогенности субстанций могут быть использованы при условии их валидации.
4.3.4. Системная токсичность
Адъюванты различных классов могут быть систематизированы по их способности вызывать токсические эффекты в различных органах. Система введения препарата должна быть разработана в соответствии с назначаемыми дозами, включая интервал при повторном введении, который предполагается при клиническом использовании.
При проведении токсикологических исследований должны проводиться патоморфологические и гистологические исследования следующих органов и тканей:
сердце, легкие, мозг, печень, почки, репродуктивные органы и др.;
кожа (при подкожном пути введения);
органы иммунной системы: селезенка, тимус, костный мозг, лимфатические узлы (локальные и отдаленные от места введения).
Полное обследование органов и тканей рекомендуется в случае использования новых адъювантов, которые не изучались на этапе доклинических испытаний и не имеют опыта клинического применения.
Токсичность главным образом вызвана иммуностимулирующим эффектом адъюванта, но также нельзя исключать его прямое токсическое воздействие на другие органы, не являющиеся органом-мишенью. Диапазон доз может оставаться относительно низким, соответствуя дозам при его клиническом использовании, не достигая максимально переносимой дозы.
4.3.5. Репродуктивная токсичность.
В соответствии с программами вакцинации вакцина с адъювантом может быть предназначена также для женщин детородного возраста, поэтому важно изучить ее репродуктивную токсичность. Более того, вакцина может назначаться беременным женщинам для предупреждения инфекционного заболевания плода путем обеспечения пассивной иммунизации. Исследование репродуктивной токсичности должно быть выполнено с адъювантом, предназначенным для использования в данном типе вакцин. Программа исследования должна быть составлена с учетом предполагаемой схемы применения вакцины. Иммунный ответ на бустерную дозу может отличаться от первичного ответа, в связи с этим первую дозу вакцины вводят до наступления беременности, а последующие бустерные дозы - во время беременности.
4.3.6. Генотоксичность.
Адъюванты могут быть биологического или синтетического происхождения. В соответствии с требованиями глав 5.3 - 5.4 настоящих Правил для биотехнологических продуктов общие требования к исследованию генотоксичности адъювантов биологического происхождения не всегда соответствуют требованиям к тем исследованиям, которые должны быть проведены для адъювантов иного происхождения. Для синтетических адъювантов исследования должны быть проведены по стандартной схеме, любые отклонения должны быть научно обоснованы.
4.3.7. Канцерогенность.
Поскольку адъюванты предназначены для введения лишь несколько раз в жизни в низких дозах, риск прямой индукции опухолей этими соединениями незначителен. Кроме того, действие адъювантов направлено на стимуляцию иммунной системы, они не являются иммунодепрессантами общего действия, что снижает риск спонтанного образования лимфоидных опухолей. Таким образом, исследования канцерогенности не требуются.
4.3.8. Комбинация адъювантов.
Использование субстанций с иммуномодулирующими свойствами совместно с адъювантами, оптимизирующими процесс презентации антигена, может сопровождаться повышением активности адъюванта. Соответствующие исследования токсичности должны быть выполнены для обеспечения безопасности использования комбинации адъювантов в дополнение к исследованиям по каждому индивидуальному компоненту. Исследования токсичности с отдельными компонентами должны быть выполнены и включены в предварительные исследования. Исследование конечной комбинации должно быть выполнено в соответствии с требованиями GLP.
4.4. Токсичность адъюванта в сочетании с антигеном
Доклинические аспекты изучения безопасности комбинации адъюванта с предполагаемым антигеном должны проводиться в соответствии с требованиями соответствующих рекомендаций Союза. Особое внимание следует уделять аспектам, изложенным в настоящем разделе главы.
4.4.1. Местная переносимость.
Инъекции антигенов в сочетании с адъювантами могут вызывать более выраженные местные реакции, чем реакции после применения только 1 адъюванта. Следует изучить оптимальное соотношение дозы адъюванта и антигена с точки зрения соотношения пользы и риска.
4.4.2. Изучение токсичности многократного введения (хронической токсичности).
Схема дозирования при исследовании хронической токсичности должна быть составлена в соответствии со схемой клинического применения. Чтобы установить безопасность схемы многократного введения (при котором увеличивается интенсивность иммунного ответа), количество введений при исследовании токсичности должно быть больше, чем планируется при вакцинации человека.
4.4.3. Характеристика иммунного ответа.
Минимальные требования к проведению исследований иммуногенности на доклиническом этапе включают в себя необходимость изучения следующих аспектов:
оценка соотношения "доза - эффект" путем исследования эффекта комбинации различных доз адъюванта с различными дозами антигена вакцины;
сравнительные исследования для установления эффекта нового адъюванта в сравнении только с антигеном вакцины или адъюванта с уже известным адъювантом.
Тип и интенсивность иммунного ответа (гуморальный или клеточный) определяют эффективность вакцины.
Тип иммунного ответа, формирующийся на один и тот же антиген вакцины, может отличаться в экспериментах на животных и у человека. Поэтому вопрос об экстраполяции данных, полученных в исследованиях на животных, требует специального рассмотрения и должен решаться с большой осторожностью. С другой стороны, до начала клинических исследований следует представить обоснованную концепцию, полученную в ходе доклинических исследований.
Если возможно, дальнейшие исследования должны быть направлены на более детальное изучение механизмов иммунологического действия нового адъюванта на соответствующих моделях животных (см. подраздел 4.1. настоящей главы).
Рациональный выбор комбинации адъювантов должен базироваться на экспериментальных данных.
5. Клинические исследования
5.1. Введение
Включение адъюванта в состав вакцины должно быть обосновано. В обосновании необходимо представить доказательства повышения иммунного ответа без увеличения местных и системных побочных реакций.
При проведении клинических исследований необходимо показать, что количество адъюванта, включаемого в вакцину, вызывает усиление иммунного ответа на антиген вакцины, что повышает эффективность вакцинации, а также повышает уровень безопасности применения препарата. Включение адъюванта в состав комбинированной вакцины должно усилить иммунный ответ хотя бы на 1 антиген без развития отрицательного влияния на иммунный ответ на другие антигены вакцины. Особое внимание следует обратить на возможность развития и повышение частоты и (или) тяжести побочных реакций в результате включения адъюванта в вакцину. Поэтому связанный с адъювантом риск не должен превышать потенциальную пользу применения препарата в результате повышения иммунного ответа.
В настоящем разделе рассматриваются следующие вопросы:
проведение клинических исследований при включении новых адъювантов в новые препараты, которые не зарегистрированы, и (или) зарегистрированные профилактические вакцины;
клинические исследования проводятся при внесении любых изменений (удаление, добавление и (или) замена) содержания адъюванта в зарегистрированных вакцинах.
Общие принципы, указанные в настоящем разделе, применяются как для вакцин, содержащих 1 антиген, так и для комбинированных вакцин вне зависимости от пути введения. Оценка некоторых особенностей характеристики иммунного ответа проводится в процессе изучения безопасности и эффективности препарата. Следует обратить внимание, что понятие "известный адъювант" применяется к вакцинам любого состава - к зарегистрированным вакцинам, содержащим 1 или несколько антигенов.
Особенности различных программ исследований связаны со следующими положениями:
новые вакцины - включение 1 или нескольких новых или известного адъюванта в новую вакцину для повышения иммунного ответа на 1 или несколько антигенов, проявляющегося в усилении эффективности вакцинации;
внесение изменений в ранее зарегистрированные вакцины - изменения в зарегистрированные вакцины вносятся для усиления или регулирования иммунного ответа и (или) для повышения безопасности их применения. В некоторых случаях включение адъюванта в вакцину проводится с целью снижения необходимого количества антигена без изменений антигенных свойств вакцины (например, вакцина против пандемического гриппа). Изменения вносятся:
при включении в препарат одного или нескольких новых или известных адъювантов;
при повышении количества адъюванта;
при снижении количества или удаления из препарата одного или нескольких адъювантов (без их замены);
при замене одного или нескольких адъювантов на новые или известные адъюванты.
5.2. Предварительные исследования
Если в качестве адъюванта используется новое вещество или разработан новый состав, то в предварительных исследованиях необходимо установить эффективность адъюванта и влияние на тип иммунного ответа на антиген, который предполагается включить в этот комплекс. Если планируется использовать в вакцине более 1 адъюванта, то необходимо проведение исследований для оценки влияния комбинации адъювантов на иммунный ответ на антиген. Кроме того, при создании комбинированных вакцин, в которых содержится более 1 из элементов комплекса адъювант - антиген, необходимо изучение активности каждого адъюванта по отношению к введенным антигенам.
5.2.1. Влияние адъювантов на иммунный ответ.
Исследование по оценке влияния адъюванта на иммунный ответ должно включать в себя изучение иммунного ответа при введении антигена без адъюванта, который должен содержаться в готовой вакцине, и антигена с адъювантом (адъювантами). При разработке комбинированных вакцин достаточно исследований, в которых следует провести сравнение иммунного ответа на комбинацию антигенов без адъюванта и комбинацию антигенов вместе с каждым из адъювантов. В процессе проведения таких ограниченных предварительных исследований особое внимание следует уделять изучению безопасности.
Обычно подобные исследования проводятся на небольшой популяции здоровых лиц. В случае если вакцина предназначена для введения новорожденным, маленьким детям или лицам старческого возраста, она должна быть изучена в соответствующей возрастной популяции при отсутствии принципиальных противопоказаний для проведения таких исследований после проведения исследований в популяции взрослых пациентов.
Исследование должно включать в себя комплексную оценку всех возможных потенциальных последствий влияния адъюванта на иммунный ответ на все антигены, которые включены в состав разрабатываемой вакцины. Кроме того, необходимо изучение иммуногенных свойств самого адъюванта. Набор тестов при изучении иммуногенности зависит от происхождения и особенностей строения изучаемых антигенов и адъювантов. Дополнительно следует учитывать результаты изучения иммуногенности на экспериментальных моделях при изучении механизма действия адъювантов.
При оценке гуморального звена иммунитета необходимо не только определение уровня антител, но и характеристика антител (нейтрализующие, опсонирующие или бактерицидные антитела), исследования проводятся с использованием международного стандарта (стандарт ВОЗ или его эквивалент). При характеристике антител, которые вырабатываются в ответ на введение вакцины, необходимо определить, к какому классу и подклассу принадлежат антитела, определить свойства иммуноглобулинов класса A (IgA) (циркулирующие в крови или секреторные) и охарактеризовать другие свойства антител, например, авидность.
Оценка клеточного звена иммунитета проводится по определению антигенспецифического T-клеточного ответа, который включает в себя характеристику Th1, Th2, T-регуляторных клеток и определение уровня соответствующих цитокинов. Спектр исследуемых показателей должен быть обоснован и может быть не ограничен указанными исследованиями. Диапазон проводимых анализов с обоснованием каждого исследования следует привести в регистрационном досье.
При использовании нового адъюванта необходимо изучить безопасность применения адъюванта с антигеном на доклиническом этапе. Аналогичные материалы по доклиническому изучению должны быть представлены, если изменяется доза (на более высокую дозу) или применяется новый путь введения адъюванта, который ранее не применялся. В случае если имеется потенциальная угроза кумуляции адъюванта в организме, необходима оценка фармакокинетических показателей в клинических исследованиях. Необходимость в проведении клинических испытаний только адъювантов должна быть обсуждена с представителями уполномоченных органов государств-членов.
5.2.2. Исследование подбора дозы.
Необходимо представить убедительные доказательства (достаточное количество данных), что соотношение количества адъюванта и антигена, выбранное для дальнейшего исследования, является оптимальным для развития иммунного ответа на антиген с минимальным риском развития нежелательных явлений. Основной целью при составлении большинства комбинаций адъювант - антиген является использование как можно меньшего количества 1 или обоих компонентов, чтобы достигнуть необходимого удовлетворительного иммунного ответа с минимальным уровнем нежелательных явлений. Предварительное количественное содержание адъюванта и антигена в комбинированном препарате определяется на основании исследований, представленных в разделе 5.2.1. настоящих Правил, которые могут проводиться одновременно с исследованием по подбору доз.
Объем исследований по подбору доз зависит от свойств предполагаемого готового препарата. Например, если предполагается использование адъюванта в дозах, которые применяются как минимум в 1 зарегистрированном препарате, то основное внимание при исследовании должно быть направлено на характеристику применения разных доз антигена. Однако, если планируется включение адъюванта в состав препарата, доза антигена или комбинации антигенов в котором используется в 1 или нескольких зарегистрированных препаратах, то основное внимание должно быть уделено характеристике количества адъюванта. В случае использования в препарате нового адъюванта и (или) нового антигена (отдельно или в комбинации) необходимо проведение более широких исследований по подбору доз.
Оптимальным является проведение исследования в популяции лиц, которым показана вакцинация данными антигенами (целевая популяция). Однако в том случае, если трудно подобрать необходимую популяцию, исследование подбора дозы может быть проведено в популяции, для которой отсутствуют показания к назначению вакцины. В случае если исследование подбора дозы не позволяет установить 1 дозу комплекса антиген - адъювант и не для 1 препарата, то возможно будет необходимым проведение подтверждающих исследований для ряда параметров. При этом желательны исследования по характеристике иммунного ответа на подобранную комбинацию антиген - адъювант по крайней мере на субъектах, включенных в подтверждающие испытания.
5.3. Подтверждающие испытания
5.3.1. Общие выводы.
Клинические исследования в основном являются рандомизированными, двойными слепыми и контролируемыми. Дизайн исследований зависит от свойств комплекса антиген - адъювант. При модификациях, которые касаются изменения содержания антигена в зарегистрированных вакцинах, должна проводиться оценка иммунного ответа для подтверждения его протективной направленности.
Необходимо представление данных изучения иммуногенности, если при проведении исследований не было получено подтверждение протективной направленности иммунного ответа.
Эти исследования должны быть проведены в популяции пациентов, для лечения или профилактики которых препараты разработаны. Если данные исследования должны включать в себя широкий возрастной диапазон пациентов, необходимо отдельно проводить исследования в группах, различающихся по возрастному составу. Например, если установлено усиление иммунного ответа на введение антигена в отдельной возрастной группе, необходимо проводить изучение иммуногенности в различных по возрастному составу группах пациентов.
5.3.2. Возможные варианты программы исследования.
5.3.2.1. Новые вакцины с новым или известным адъювантом.
Для новых вакцин (вакцин, содержащих антигены, не описанные в Фармакопее Союза или нормативных документах ВОЗ, вакцин, в которых использован новый конъюгат для известного антигена, или любых новых комбинаций известных и (или) новых вакцин) применяются требования, которые не рассматриваются подробно в настоящих Правилах.
5.3.2.2. Изменение содержания адъюванта в зарегистрированной вакцине
Необходимо проведение как минимум 1 подтверждающего исследования для обоснования изменений содержания адъюванта. Дизайн исследования зависит от того, с какой целью осуществляется изменение.
Дизайн исследований при внесении изменений для повышения эффективности препарата.
Если основной целью является повышение иммунного ответа на 1 или несколько антигенов или прямое влияние иммунного ответа на предполагаемый конечный эффект, программа исследования должна быть разработана для доказательства превосходства измененного препарата над существующим зарегистрированным препаратом. В процессе проведения исследований комбинированных препаратов должно быть доказано превосходство иммунного ответа модифицированного препарата по крайней мере к 1 из антигенов. Вторичной целью данных исследований должна быть демонстрация того, что иммунный ответ на другие антигены, которые входят в состав модифицированного препарата, не хуже. Демонстрация доказательства не меньшей активности по отношению к другим антигенам может быть использована только в том случае, если предварительно доказано превосходство адъюванта по показателям эффективности.
Если предполагается использование адъюванта в большей дозе, чем в ранее зарегистрированном препарате, или изменение способа введения, необходимы дополнительные исследования по оценке безопасности. Необходимость проведения таких исследований должна быть обсуждена со специалистами национального регуляторного органа до подачи заявления для регистрации модифицированного препарата.
Дизайн исследований при внесении изменений для повышения безопасности препарата.
Если основной целью разработки вакцины является повышение безопасности, в программу клинических испытаний необходимо включить исследования, которые должны доказать, что иммунный ответ на антиген модифицированной вакцины имеет не меньшую эффективность, чем ответ на каждый антиген зарегистрированной вакцины. Критерии для оценки не меньшей эффективности должны быть обоснованы при характеристике свойств антигена. Следует ожидать, что результаты данного исследования позволят обосновать улучшение показателей безопасности.
Данные по безопасности должны свидетельствовать о достоверном повышении ответной реакции, учитывая особенности адъюванта и антигена. В некоторых случаях может быть целесообразным обратить внимание на возможность развития иммунно-опосредованных реакций. В любом случае отношение "риск - польза" при применении модифицированных препаратов должно быть таким же благоприятным, как и для зарегистрированного препарата.
Пострегистрационные наблюдения должны проводиться во всех случаях внесения изменений в состав зарегистрированной вакцины, содержащей адъювант.
5.3.3. Статистическая обработка результатов исследования.
Оцениваемые показатели и методы, используемые для проведения статистического анализа, должны быть представлены в протоколе клинических исследований. Объем выборки должен быть достаточным для достоверного обоснования результатов исследований. Необходимо заранее представить и обосновать применение критериев не меньшей активности и обосновать значение допустимого предела при их применении. При планировании клинических испытаний необходимо учесть возможные проблемы, связанные с объемом выборки. При составлении программы исследований могут быть использованы научные рекомендации и требования актов, входящих в право Союза.
Глава 17. Клинические исследования препаратов
для специфической иммунотерапии аллергических болезней
1. Введение
Настоящая глава содержит указания для подготовки программы исследования эффективности и безопасности препаратов аллергенов (препаратов на основе экстрактов аллергенов, рекомбинантных аллергенов, модифицированных аллергенов и др.), разработанных для специфической иммунотерапии аллергических заболеваний (например, длительное лечение и коррекция таких аллергических заболеваний, как риноконъюнктивит и аллергия на яды насекомых).
Во всем мире наблюдается увеличение количества аллергических заболеваний, таких как аллергический ринит (риноконъюнктивит), аллергическая астма, пищевая аллергия, обусловленное факторами окружающей среды, генетической предрасположенностью и естественным развитием аллергических иммунных реакций. Контакт организма с аллергенами окружающей среды (из пыльцы растений, клещей, перхоти животных, грибов, яда насекомых и продуктов питания), может привести к развитию иммуноглобулин E-зависимой (IgE-зависимой) сенсибилизации, и при повторном контакте с аллергенами могут появиться симптомы заболевания.
Кроме симптоматического лечения, основанного на фармакотерапии, важную роль играют такие длительные мероприятия, как профилактика и иммуномодулирующая терапия. Специфическая иммунотерапия препаратами аллергенов основана на многократном введении больным аллергенов для активации иммуномодулирующих механизмов для устойчивого снижения симптомов аллергии, потребности в лекарственных средствах и повышения качества жизни в период контакта с естественными аллергенами. Специфическая иммунотерапия аллергии, вызванной ингаляционными аллергенами, приводит к изменению (модификации) обусловленных заболеванием эффектов. IgE-зависимая пищевая аллергия является причиной развития тяжелой анафилаксии, в том числе с летальным исходом, при этом ее лечение пока отсутствует.
Механизмы клинической эффективности специфической иммунотерапии до конца не изучены, установлено, что иммунотерапия изменяет течение аллергического процесса. Например, под влиянием специфической иммунотерапии происходит повышение уровня аллергенспецифичных блокирующих IgG (IgG4 и IgA) антител и снижение или подавление синтеза аллергенспецифичных IgE антител. При этом наблюдается изменение состава и активности эффекторных клеток, в том числе тучных клеток, эозинофилов и базофилов. Данные эффекты обусловлены изменениями ответа на аллерген T-лимфоцитов. При преобладании активности Th2-типа иммунного ответа, который сопровождается увеличением синтеза интерлейкина-4 (IL) и IL-5 при аллергии, под влиянием иммунотерапии происходит сдвиг в сторону повышения активности Th1-типа ответа и синтеза интерферона-гамма и IL-2. Это вызывает стимуляцию T-регуляторных клеток и увеличение синтеза IL-10 и TGF-бета.
Данные изменения под влиянием специфической иммунотерапии приводят к подавлению индуцированного аллергеном T-клеточного ответа в коже и легких и подавлению на длительный срок симптомов заболевания, которые могут развиться после прекращения специфической иммунотерапии. Тем не менее, до сих пор механизм специфической иммунотерапии изучен не полностью, и в настоящее время ни одно из указанных изменений системы иммунитета не может быть использовано для прогнозирования клинического исхода. В то же время данные изменения системы иммунитета коррелируют с клиническими показателями и должны учитываться при клиническом анализе.
Для изучения новых препаратов для специфической иммунотерапии используется большое количество различных схем проведения клинических исследований, которые различаются между собой по исследуемым дозам, продолжительности исследования, критериям включения, конечным точкам, методам анализа результатов и контролю параметров окружающей среды.
В настоящей главе представлены указания для разработки плана изучения препаратов, предназначенных для специфической иммунотерапии, совершенствования подходов для оценки и сравнения результатов.
В настоящей главе представлены требования для проведения клинического изучения специфической иммунотерапии при любой локализации аллергического процесса (например, в области верхних и нижних дыхательных путей, глаз, при системной реакции) с использованием аллергенов, полученных из любого сырья (например, из пыльцы, клещей, шерсти животных, грибов, яда насекомых, продуктов питания), любых аллергенных препаратов (например, экстрактов, очищенных аллергенов, модифицированных аллергенов, адсорбированных аллергенов) и для всех способов введения препарата (например, подкожного, подъязычного). Данные указания не распространяются на такие заболевания, как атопическая экзема (дерматит) или астма, при которых аллерген не является причиной развития болезни.
2. Сфера применения
Настоящая препаратспецифичная глава содержит доклинические и клинические требования к исследованию лекарственных препаратов для специфической иммунотерапии аллергических заболеваний.
3. Связь с другими главами
В главах 13 и 14 настоящих Правил содержатся общие указания по разработке биологических лекарственных препаратов.
4. Основные указания
4.1. Характеристика и выбор пациентов
4.1.1. Диагностика.
В исследование должны быть включены больные, которые имеют подробный аллергологический анамнез заболевания.
Диагноз IgE-опосредованных заболеваний устанавливается на основе критериев, представленных в международных рекомендациях, в анамнезе должна быть информация об обострении сезонного аллергического ринита (риноконъюнктивита) или аллергической астмы не менее 2 лет подряд и один год обострения, если аллергическое заболевание вызвано круглогодичным аллергеном. Анамнез больных с инсектной аллергией должен содержать подробное описание симптомов аллергии после укуса перепончатокрылых согласно классификации.
4.1.2. Выбор пациентов.
У больных аллергическими заболеваниями дыхательных путей может определяться разное количество аллергенов, которые вызвали сенсибилизацию. Очень мало больных, у которых определяется сенсибилизация к 1 аллергену (моносенсибилизация), в основном определяется полисенсибилизация к нескольким аллергенам одного вида или полисенсибилизация к нескольким аллергенам разного вида. В связи с этим очень сложно подобрать группу больных, у которых определяется моносенсибилизация. Поэтому в исследование необходимо включать больных, у которых определяется ограниченное (минимальное) количество аллергенов, которые вызвали сенсибилизацию. При этом необходимо документально указать причинные аллергены и представить оценку других аллергенов, к которым сенсибилизирован пациент. Определение аллергенов проводится в кожных тестах и при определении аллергенспецифичных IgE антител (с использованием методов количественного определения IgE, которые должны быть валидированы). Все определения аллергенов должны быть документально подтверждены. Кроме того, необходимо определение аллергенов, иммунотерапия против которых не будет проводиться, но которые могут повлиять на результаты исследований. Данные исследования необходимы при иммунотерапии сезонного аллергического ринита (риноконъюнктивита), вызванного пыльцой или грибами, и круглогодичного аллергического ринита (риноконъюнктивита), вызванного клещами или шерстью животных. При наличии у больного сенсибилизации к сезонным и круглогодичным аллергенам могут возникнуть дополнительные трудности. Прежде всего, следует учесть, что не все аллергены, которые вызвали сенсибилизацию, имеют клиническое значение. Для получения объективных результатов изучения иммунотерапии рекомендуется не включать в одно исследование больных, у которых определяется клинически значимая сенсибилизация к аллергенам разных сезонов года, и больных, у которых временные периоды обострений могут накладываться друг на друга. Также не рекомендуется включать в исследование больных, у которых определяется сенсибилизация к круглогодичным аллергенам и у которых может развиться обострение в период активности сезонного изучаемого аллергена. При изучении круглогодичных аллергенов в исследование могут быть включены больные, у которых отсутствует сенсибилизация к другим круглогодичным аллергенам, которая имеет клиническое значение. При изучении круглогодичных аллергенов среди больных, у которых определяется клинически значимая сенсибилизация к сезонным аллергенам, оценка эффективности не должна проводиться в период, который соответствует активности сезонного аллергена.
У больных аллергией к ингаляционным аллергенам, которые планируется включить в исследование, до начала исследования должен определяться минимальный уровень соответствующих симптомов заболевания. Оптимальной является характеристика симптомов при проведении оценки исходного состояния больного, а не представление в баллах симптомов, которые были раньше. В исследование не допускается включение больных, которые получали иммунотерапию с использованием изучаемого аллергена, перекрестнореагирующего аллергена или любого аллергена, для которого имеются основания для невключения, в течение последних 5 лет.
Больные с высоким риском развития анафилактических реакций на укус насекомых, которых планируется включить в исследование иммунотерапии инсектными аллергенами, должны быть обследованы на мастоцитоз. В связи с высоким риском развития анафилактических реакций на укус насекомых при изучении иммунотерапии аллергеном из яда насекомых необходимо учитывать наличие мастоцитоза при оценке эффективности и безопасности.
Выбор критериев включения в исследование осуществляется с учетом возраста, пола, особенности и тяжести заболевания, наличия сопутствующих заболеваний, предыдущей иммунотерапии и др. К критериям исключения могут быть отнесены такие факторы, как необходимость приема препаратов, наличие других заболеваний и др. Выбор критериев должен быть обоснован и указан в протоколе исследования, чтобы была возможность оценить достоверность результатов.
4.1.3. Сопутствующие заболевания.
Очень часто у одного больного встречается ринит (риноконъюнктивит) и астма. При изучении эффективности специфической иммунотерапии ринита (риноконъюнктивита) в исследование могут быть включены больные с сопутствующей астмой для получения данных о безопасности иммунотерапии и оценке влияния препарата на астму. Несмотря на это отдельно проводится оценка эффективности у больных с астмой, особенности терапии астмы должны быть соблюдены.
4.2. Лекарственные средства, не являющиеся аллергенами
4.2.1. Совместная терапия.
Все препараты, которые принимают больные (в том числе без рекомендаций врача), должны быть указаны в соответствующих документах, при этом необходимо определить препараты, которые могут повлиять на результаты исследования, и исключить их прием. Кроме того, необходимо определить, какие препараты рекомендуется назначать для неотложной терапии.
4.2.2. Препараты неотложной терапии.
При проведении клинических исследований должны быть предусмотрены препараты неотложной терапии. Критерии для назначения больному препаратов неотложной терапии (в зависимости от симптомов и (или) тяжести симптомов) необходимо указать в протоколе исследования. Если у больного астматический статус контролируется препаратами, то они должны рассматриваться как средства неотложной терапии. Каждый случай применения препарата неотложной терапии должен быть занесен в дневник пациента. Не рекомендуется переходить на прием сильнодействующего препарата после окончания лечения, предпочтительным является применение препаратов неотложной терапии с коротким фармакодинамическим эффектом. При оценке эффективности необходимо учитывать, что препараты неотложной терапии могут оказывать влияние на результаты исследования.
4.3. Клинические исследования
4.3.1. Предварительные исследования.
Фаза I классической схемы клинических исследований на здоровых добровольцах не подходит для оценки аллергенов для специфической иммунотерапии, так как не позволяет оценить эффективность и безопасность препарата на этом этапе. Лица, у которых отсутствует гиперчувствительность, не могут реагировать, как сенсибилизированные больные, и у них не определяются риски, которые наблюдаются у больных аллергией. Таким образом, изучение аллергенных препаратов проводится только в популяции больных аллергией, а оценка местнораздражающего действия препарата возможна с участием здоровых добровольцев. Для получения предварительных данных о безопасности и переносимости максимальной переносимой дозы и выбора подходящей схемы повышения дозы препарата (эскалации) необходимо изучение разных доз препарата. Необходимость достижения поддерживающей дозы зависит от схемы повышения дозы, что должно быть продемонстрировано в соответствующих исследованиях.
4.3.2. Исследования выбора дозы.
После определения диапазона переносимых доз необходимо провести изучение зависимости "доза - эффект". Данные исследования могут быть выполнены при проведении нескольких краткосрочных неконтролируемых исследований (например, 2 - 4 месяца). В качестве первичных конечных точек могут быть использованы провокационные тесты (например, конъюнктивальной, назальной или бронхиальной провокации или определение экспозиции аллергена в камере) и клинические показатели. Лабораторные показатели, имеющие клиническое значение при проведении длительных исследований (специфические антитела, T-клеточный ответ, цитокины) не могут быть использованы для оценки терапевтической дозы, так как на данном этапе исследования они не определяются и не коррелируют с клиническим ответом на введение препарата.
4.3.3. Исследование фармакокинетики и фармакодинамики.
Оценку фармакокинетических свойств препаратов для специфической иммунотерапии провести невозможно, так как в плазме нельзя определить концентрацию действующего вещества данных препаратов.
Для аллергенных препаратов невозможно проведение обычных исследований фармакодинамики. Однако, чтобы продемонстрировать влияние специфической иммунотерапии на систему иммунитета, можно оценить динамику показателей иммунитета (например, изменение уровня аллергенспецифичных IgG, T-клеточного ответа и (или) синтеза цитокинов) и (или) оценка ответа органа (ткани) (например, провокационные тесты). Данные исследования могут быть выполнены в рамках дальнейших исследований препаратов.
4.3.4. Подтверждающие исследования.
Дизайн исследования.
Подтверждающие исследования препаратов для иммунотерапии проводят в рандомизированных, плацебоконтролируемых, двойных слепых исследованиях. Применение критериев не меньшей эффективности не позволяет объективно оценить результаты исследований, так как ответы на применение аллергенов колеблются в широком диапазоне и могут быть непредсказуемыми.
В связи с этим рекомендуется проведение сравнительных исследований с плацебо - объектом сравнения. Учитывая, что при проведении специфической иммунотерапии часто встречаются местные побочные реакции, клинические исследования с использованием препарата плацебо с гистамином должны быть слепыми.
Однако при иммунотерапии аллергии на укус насекомых использование плацебо в контрольной группе является неэтичным. В данном случае в качестве контроля может использоваться зарегистрированный препарат для лечения инсектной аллергии. При этом необходимо проведение слепого исследования, которое проводится с использованием одинаковых схем и путей введения изучаемого и контрольного препаратов, или использование другого дизайна исследования (например, исследование с двойной маскировкой). В особых случаях, когда приходится использовать проведение исследований не меньшей эффективности, основное внимание необходимо уделить выбору чувствительных методов оценки сенсибилизации. Для этого рекомендуется обратиться в уполномоченные органы для обсуждения дизайна исследования.
В клинические исследования специфической иммунотерапии должны быть включены только те больные, которые имеют хотя бы минимальный уровень симптомов (в соответствующий период времени) перед началом исследования. На данной стадии можно учитывать симптомы, которые отмечались ранее (основанные на анамнезе), но их не рекомендуется использовать в дальнейшем при учете результатов или в сравнительных исследованиях. Преимущественным является подход, связанный с основным периодом исследования, что позволяет оценить симптомы заболевания и воздействие препарата. При этом необходимо учитывать, что аллергены, особенно пыльцевые, могут воздействовать непредсказуемо или с широкими вариациями, что снижает информативность результатов, полученных от указанных подходов. Для включения в исследование больного с необходимым минимальным уровнем симптомов можно использовать провокационные тесты с титрованием.
При исследовании сезонной аллергии обязательным является указание в протоколе того уровня пыльцы в атмосфере, при котором возможно начало оценки симптомов заболевания, и основного периода наблюдения.
При проведении клинических исследований круглогодичных аллергенов необходимо минимизировать различия в уровне аллергенов, которые определяются внутри помещений. Поэтому, если планируется проведение санитарных мероприятий, они должны быть закончены до начала клинического исследования, оценку симптомов заболевания необходимо проводить после окончания санитарных мероприятий. В процессе проведения исследований никакие санитарные мероприятия не проводятся. Кроме того, вариации уровня домашних аллергенов конкретных больных, особенно в период оценки симптомов, рекомендуется отразить в документации.
Продолжительность исследования.
Основной целью специфической иммунотерапии является получение стойкого эффекта, вызванного изменениями в системе иммунитета, что может быть доказано в долгосрочных исследованиях. Тем не менее достоверные результаты эффективности специфической иммунотерапии при аллергическом рините (риноконъюнктивите) могут быть получены после оценки за 1 сезон пыления растений или за 1 или 2 контрольных периода для круглогодичной аллергии. В зависимости от продолжительности изучения эффективности возможны следующие варианты исследований:
оценка лечения аллергических симптомов - краткосрочные клинические исследования, показывающие эффективность в первый сезон пыления растений после начала специфической иммунотерапии или при круглогодичной аллергии после нескольких месяцев лечения;
оценка достижения устойчивого клинического эффекта - обеспечение значительной и клинически значимой устойчивой эффективности в течение 2 - 3 лет лечения.
оценка долгосрочной эффективности и изменение эффектов, вызываемых заболеванием - подтверждение достоверной и клинически значимой эффективности, определяемое годами, после лечения;
лечение аллергии - подтверждение отсутствия аллергических симптомов, определяемое годами после лечения.
На основании данных условий разрабатываются отдельные конкретные исследования.
Кроме того, долгосрочные исследования могут быть запланированы как дополнение для изучения предотвращения развития астмы и увеличения спектра аллергенов, которые стали причиной сенсибилизации. Если в исследовании запланированы более чем 1 цель, необходимо особое внимание уделить методологической стороне составления программы исследования (например, многоцелевое исследование). При этом не рекомендуется использование данных промежуточных исследований, так как они могут оказать влияние на результаты продолжающегося исследования.
Оценка конечных точек проводится в каждый сезон для сезонного аллергена и несколько раз в течение лечения или в конце наблюдения для круглогодичного аллергена. Повышение эффективности в процессе лечения будет указывать на эффект препарата, но не позволяет сделать окончательный вывод об этом, так как эффективность зависит от экспозиции аллергена, которая может быть разной в разные годы. При этом высокая эффективность в первый год лечения может привести к тому, что в следующий год не будет отмечено увеличения эффективности и непрерывное лечение может быть необходимо для получения долгосрочного стойкого эффекта. В связи с этим для оценки эффективности, особенно в периоды (годы) с низким уровнем аллергена и, соответственно, низкой (или даже нулевой) эффективностью, можно использовать провокационные тесты. При этом если концентрация аллергена, которая необходима для развития симптомов аллергии, со временем увеличивается, то это может свидетельствовать об эффективности иммунотерапии. Однако подобные тесты не могут использоваться в качестве суррогатных маркеров эффективности.
Для оценки лечения инсектной аллергии необходимо обосновать период проведения иммунотерапии. При этом необходимо учесть необходимость долгосрочной оценки эффективности.
Конечные точки.
Клинически значимые различия первичных конечных точек между исследуемой и контрольной группами должны быть описаны и обоснованы заявителем. Выбор конечных точек при проведении подтверждающих клинических исследований зависит от изучаемой патологии:
аллергический ринит (риноконъюнктивит):
первичная конечная точка:
применение препаратов неотложной терапии для купирования симптомов аллергии. Первичная конечная точка должна отражать тяжесть симптомов и применение препаратов неотложной терапии. Оценка пациентом выраженности симптомов часто используется в качестве первичного критерия эффективности при клинических исследованиях с аллергическим ринитом (риноконъюнктивитом) с аллергической астмой или без астмы. Данная оценка должна проводиться больным ежедневно в течение периода, установленного для изучения эффективности. Несмотря на то, что 4-балльная шкала для оценки симптомов не валидирована, она используется для оценки симптомов аллергии:
0 - отсутствуют симптомы (без явных симптомов (признаков) аллергии);
1 - легкие симптомы (присутствуют симптомы (признаки) слабой выраженности, которые легко переносятся);
2 - умеренные симптомы (присутствуют симптомы (признаки), которые доставляют беспокойство больному, но терпимы);
3 - тяжелые симптомы (присутствуют симптомы (признаки), которые тяжело переносятся, являются помехами в повседневной жизнедеятельности и (или) сне).
Характерными симптомами аллергического ринита (риноконъюнктивита) являются зуд в носу, чихание, насморк, заложенность носа, зуд в глазах, ощущение песка в глазах, покраснение и слезотечение.
Оценка симптомов проводится в отсутствие регулярного применения лекарственных средств. Препараты неотложной терапии оказывают различное влияние на выраженность и длительность симптомов аллергии, могут влиять на различные органы и системы, что требует клинического обоснования с учетом облегчения симптомов (уровня и длительности эффекта) и вида симптомов.
Возможны различные подходы для того, чтобы одновременно учесть применение шкалы для оценки симптомов и прием препаратов неотложной терапии, которые должны быть обоснованы, отсутствует Система для оценки эффективности, которая позволила бы скомпенсировать выраженность симптомов заболевания на фоне приема лекарственных средств, отсутствует. Необходимо проводить собственную разработку сбалансированной и валидированной системы оценки эффективности.
Одним из подходов решения данной проблемы является объединение показателей, характеризующих симптомы аллергии и используемых для учета приема лекарственных средств. Влияние каждого фактора должно быть обосновано. Все исследования с использованием данного комбинированного подхода должны опираться на данные, полученные у больных, ответивших на лечение (например, больных у которых оценка с использованием комбинированного показателя была ниже уровня, определенного для учета ответа). Альтернативный подход заключается в объединении шкалы для учета симптомов и применение препаратов неотложной терапии, в качестве показателя используется количество дней без развития симптомов и приема препаратов неотложной терапии.
При необходимости и клинической обоснованности могут быть использованы другие конечные точки. В случае если имеются ограничения для оценки эффективности в определенный период времени, который определен для оценки первичной конечной точки, это необходимо отразить в протоколе.
Вне зависимости от выбора критерия для оценки эффективности показатель первичной конечной точки должен быть выражен в единицах и иметь клиническую значимость. Для оценки эффективности представление только статистически достоверных результатов не всегда бывает достаточным;
вторичные конечные точки:
в качестве вторичных конечных точек могут быть использованы частота возникновения симптомов аллергии, частота приема препаратов, индивидуальная оценка симптомов, оценка качества жизни с помощью опросников (HRQoL), выраженность симптомов аллергии по визуальной шкале (VAS), количество дней без симптомов, оценка пациентом и врачом улучшения общего состояния больного.
Использование результатов провокационных тестов (кожа, глаза, нос, бронхи, стимуляция аллергеном в камере) и данных объективного исследования (например, определение уровня аллергенспецифичных IgE и IgG антител, цитокинов и других маркеров воспаления) позволяет получить дополнительную информацию, но не может использоваться в качестве суррогатных маркеров эффективности и заменить качественную и количественную оценку клинических симптомов аллергии. При проведении провокационных тестов учет результатов рекомендуется проводить с использованием объективных методов оценки (например, риноманометрии). Перспективным показателем оценки эффективности является провокационный тест в аллергенных камерах, однако результаты изучения провокационного теста должны быть подтверждены при сопоставлении с результатами клинической оценки симптомов при естественной экспозиции аллергена. Кроме того, провокационные тесты должны быть подтверждены оценкой клинических симптомов в период до цветения растения и в сезон цветения (влияние воспаления), исследования в этот период могут соответствовать первичной конечной точке. Тем не менее, провокационные тесты с проведением экспозиции в аллергенной камере могут быть важным показателем, особенно при проведении долгосрочных исследований в течение нескольких лет иммунотерапии поллиноза, в которых оценить количество и тяжесть симптомов сложно из-за низкого уровня концентрации пыльцы в атмосфере.
Оценка профилактической эффективности специфической иммунотерапии бронхиальной астмы и появлений сенсибилизации к новым аллергенам может быть проведена с использованием кожного прик-теста;
аллергия к укусам насекомых:
эффективность иммунотерапии инсектной аллергии учитывается в контролируемом провокационном тесте с аллергеном яда насекомых. Оценка результатов проводится согласно принятой системы классификации.
Вопросы методологии.
При составлении плана проведения клинических исследований специфической иммунотерапии необходимо руководствоваться соответствующими международными рекомендациями (ICH) и рекомендациями Союза.
В целях избежания внесения изменений в протокол, особое внимание при подготовке программы исследований следует уделить методам оценки результатов. При отсутствии данного раздела будет невозможно определить достоверность изменения показателей при последующих определениях. Особое внимание следует уделить обработке результатов при наличии пропущенных значений и оценке результатов, включающих в себя много показателей, при анализе показателей первичных и вторичных конечных точек. Необходимо указать методы оценки результатов в подобных случаях.
В исследованиях, в которых определенную роль играет окружающая среда (например, сезонный риноконъюнктивит), должны быть описаны методы оценки экспозиции аллергенов, которые также необходимо отразить в разделе, посвященном оценке результатов.
4.3.5. Пищевая аллергия.
Проведение клинических исследований специфической иммунотерапии IgE-опосредованной пищевой аллергии имеет ряд ограничений.
Эталоном диагностики и оценки эффективности лечения пищевой аллергии является проведение двойного слепого плацебоконтролируемого пищевого теста (DBPCFC). В связи с тем, что при применении пищевых аллергенов высока вероятность развития аллергических реакций и имеется небольшой опыт проведения специфической иммунотерапии данной аллергии, для проведения клинических испытаний специфической иммунотерапии необходимо проведение консультаций с регуляторными центрами с учетом конкретных условий исследования.
4.3.6. Очищенные аллергены (нативные, рекомбинантные, синтетические пептиды).
К очищенным аллергенам относятся все очищенные аллергены вне зависимости от происхождения - экстракты или полученные с использованием технологии рекомбинантной ДНК. Кроме того, химически синтезированные пептиды, строение которых соответствует аллергену, также относятся к очищенным аллергенным компонентам. Если проводится исследование очищенных аллергенов, необходимо учитывать дополнительные проблемы.
Исходные материалы для получения аллергенов могут содержать несколько соответствующих аллергенов. В зависимости от степени сенсибилизирующей способности аллергены делятся на главные ( 50 процентов связывания аллергенспецифичных IgE) и малые (< 50 процентов связывания аллергенспецифичных IgE). При этом следует отметить, что не все аллергены, которые имеются в исходном материале, известны. Пациенты, у которых установлена сенсибилизация к индивидуальному набору аллергенов, должны рассматриваться индивидуально. Использование для специфической иммунотерапии аллергенных экстрактов, которые содержат широкий спектр аллергенов, повышает вероятность того, что для лечения используется весь спектр аллергенов, которые связаны с его аллергией, даже если не все аллергены имеют отношение к его индивидуальной аллергии. Однако использование для иммунотерапии очищенных аллергенов, которые содержат меньшее количество аллергенов относительно экстракта, позволяет адекватно подобрать соответствующие аллергены и их дозы для достижения клинического эффекта. Поэтому заявитель должен обосновать выбор аллергенов и критерии отбора больных для участия в исследовании (например, оценка индивидуальной картины сенсибилизации).
50 процентов связывания аллергенспецифичных IgE) и малые (< 50 процентов связывания аллергенспецифичных IgE). При этом следует отметить, что не все аллергены, которые имеются в исходном материале, известны. Пациенты, у которых установлена сенсибилизация к индивидуальному набору аллергенов, должны рассматриваться индивидуально. Использование для специфической иммунотерапии аллергенных экстрактов, которые содержат широкий спектр аллергенов, повышает вероятность того, что для лечения используется весь спектр аллергенов, которые связаны с его аллергией, даже если не все аллергены имеют отношение к его индивидуальной аллергии. Однако использование для иммунотерапии очищенных аллергенов, которые содержат меньшее количество аллергенов относительно экстракта, позволяет адекватно подобрать соответствующие аллергены и их дозы для достижения клинического эффекта. Поэтому заявитель должен обосновать выбор аллергенов и критерии отбора больных для участия в исследовании (например, оценка индивидуальной картины сенсибилизации).
4.3.7. Перекрестно-реагирующие аллергены.
Основные понятия о перекрестно-реагирующих аллергенах (аллергенных семействах или гомологичных рядах (группах)) могут быть использованы для оценки эффективности. Для одного гомологичного ряда достаточно доказать эффективность аллергена, который выбран в качестве представителя ряда. В то же время это предполагает невозможность экстраполировать результаты изучения эффективности на другие гомологичные ряды. В случае, если положения гомологичного ряда распространяются на аллергены, которые не входят в состав этого ряда, заявитель обязан представить обоснование для включения данного аллергена в состав группы. Применение концепции гомологичных рядов к очищенным аллергенам (нативным, рекомбинантным, синтетическим) необходимо обосновать в каждом конкретном случае.
4.3.8. Аллергены, не обладающие перекрестной реактивностью.
При использовании для клинических исследований различных комбинаций аллергенов, которые получены из различных материалов, не обладающих перекрестной реактивностью (смеси экстрактов аллергенов), или различных типов аллергенов (например, очищенных аллергенов (природных, искусственных, полученных с использованием генно-инженерных технологий)) заявителю необходимо обратиться за научной консультацией в уполномоченный орган.
4.3.9. Исследования сопоставимости.
При внесении изменений в производственный процесс, которые могут повлиять на аллергенную активность препарата, необходимо проведение исследования сопоставимости. В зависимости от характера вносимых изменений сопоставимость препаратов до и после внесения изменений может быть подтверждена при сравнительном изучении in vitro физико-химических и биологических свойств по таким показателям, как состав аллергенов, активность и биологическая активность. При этом может возникнуть необходимость оценки биологической активности с использованием кожного тестирования или проведение исследований переносимости и эффективности. Для этого необходимо учитывать соответствующие нормативные требования. В зависимости от каждого конкретного случая, заявителем должно быть представлено обоснование проведения данных исследований с учетом научных рекомендаций ведущих центров.
4.3.10. Различные пути введения.
Приведенные рекомендации распространяются на все пути введения. Это не допускает экстраполяции результатов исследования одного пути введения на другой, соответствующие клинические исследования должны быть проведены для каждого пути введения. Для сравнительной оценки различных путей введения проведение двойных слепых плацебоконтролируемых исследований, как правило, не требуется.
4.4. Изучение эффективности иммунотерапии при клинических
исследованиях с участием детей
В связи с тем, что специфическая иммунотерапия используется для лечения детей, необходимо изучение эффективности и безопасности аллергенных препаратов при их применении у детей. При проведении данных исследований необходимо соблюдение всех действующих нормативных требований для проведения исследований в детской популяции (например, рекомендаций Союза и т.д.). Проведение клинических исследований с участием детей, особенно очень маленьких, имеет ряд проблем (например, оценка симптомов и использование препаратов неотложной терапии, безопасность и положительная оценка результатов исследования). Поэтому оценку эффективности необходимо проводить при проведении специальных исследований среди детей, а не в совместных исследованиях с участием взрослых и детей. Допускается проведение совместных исследований с участием подростков и взрослых.
Проведение клинических исследований проводится на основании данных указаний. Детям, которые самостоятельно не в состоянии оценить свое состояние, должны помочь родители при оценке симптомов и принятии решения для приема препаратов. Если при проведении исследований в качестве вторичной конечной точки проводится оценка качества жизни для этого необходимо использовать специальные опросники для оценки качества жизни детей, больных аллергическим ринитом (риноконъюнктивитом) или астмой.
4.5. Изучение безопасности
При проведении клинических исследований должны быть соблюдены все нормативные требования для оценки безопасности. При выявлении нежелательного явления необходимо определить его тяжесть (легкое, средней тяжести и тяжелое), провести анализ для определения его связи с изучаемым препаратом и все материалы внести в соответствующие документы. Описание нежелательных явлений необходимо проводить с использованием унифицированных кодов и терминологии. Серьезные нежелательные явления, особенно связанные с лечением, необходимо описывать очень подробно. Отдельно необходимо представить информацию об ожидаемых аллергических нежелательных явлениях. Ожидаемые аллергические нежелательные явления в зависимости от времени появления симптомов могут быть немедленного и замедленного типа (к немедленным относятся реакции, которые развиваются в течение первых 30 минут после введения препарата). В зависимости от локализации они могут быть в виде местных и системных реакций (к местным относятся реакции, которые развиваются в месте введения, а к системным - реакции, которые развиваются в местах, удаленных от места введения). Для характеристики системных аллергических реакций может быть использована классификация тяжести системных эффектов Европейской академии аллергологии и клинической иммунологии (EAACI). Среди других параметров безопасности необходимо учитывать клинические показатели жизненно-важных органов, клинический и биохимический анализ крови и анализ мочи.
Определения
В настоящей главе используются понятия, которые означают следующее:
"адсорбированные аллергены" - аллергены или экстракты аллергенов, которые адсорбированы на твердом носителе (например, алюминия гидроксиде, тирозине) для создания эффекта депонирования;
"аллерген" - 1) лекарственное средство, содержащее аллергены или производные аллергенов для лечения аллергических заболеваний; 2) молекула, способная индуцировать IgE-ответ и (или) аллергическую реакцию I типа;
"гомологичные ряды" - аллергенные экстракты, полученные из различных видов пыльцы растений, принадлежащих к разным родам или семействам, и готовые формы препаратов, полученных из данных экстрактов, которые могут быть сгруппированы в гомологичные группы в зависимости от состава, физико-химических и биохимических свойств исходного материала, перекрестной реактивности, структурной гомологии аллергенов, состава готовой формы препарата, производственного процесса и готовой формы;
"модифицированные аллергены (аллергоиды)" - химически модифицированные аллергены для снижения IgE-зависимой реактивности;
"очищенные аллергены" - аллергены, содержащие в своем составе все очищенные аллергены вне зависимости от происхождения (экстракты или полученные с использованием технологии рекомбинантной ДНК);
"схема эскалации дозы" - схема, основанная на введении постоянно увеличивающихся доз аллергена до достижения безопасной поддерживающей дозы;
"экстракт аллергена" - извлечение из исходного сырья природного происхождения, которое содержит смесь аллергенов и других молекул, не вызывающих аллергическую реакцию.
Глава 18. Производство, качество, доклинические,
клинические исследования лечебно-профилактических
препаратов бактериофагов
1. Производство бактериофагов
1.1. Введение
Бактериофаги являются вирусами бактерий, их естественными врагами и регуляторами популяции. Бактериофаги чрезвычайно широко представлены в природе во всех ареалах обитания бактерий, с оценкой общей численности в биосфере до 1030 - 32 частиц. По типу взаимодействия с бактериями они различаются на умеренные и вирулентные (литические). Умеренные фаги способны к интеграции в геном бактерии в виде профага либо могут размножаться, не вызывая гибели бактерий. В медицине их используют только в диагностических целях (например, для внутривидового типирования бактерий). Вирулентные фаги, проникая внутрь бактериальной клетки, немедленно переключают ее метаболизм на воспроизведение новых фагов, которое завершается лизисом бактерии. Литический процесс повторяется с новыми и новыми бактериальными клетками. Именно вирулентные (литические) бактериофаги обладают наибольшим терапевтическим потенциалом, поскольку только они способны к уничтожению клеток бактерий-хозяев. На этом принципе основано использование фагов при лечении и профилактике гнойно-воспалительных инфекций бактериальной природы, коррекции дисбиотических состояний. Эти препараты представляют собой стерильные очищенные фильтраты фаголизатов соответствующих видов бактерий. Они освобождены от продуктов жизнедеятельности бактерий, эндо- и экзотоксинов, продуктов фаголизиса бактериальных клеток, белковых и антигенных комплексов питательных сред.
Благодаря строгой специфичности действия бактериофаги, в отличие от антибиотиков, не угнетают нормальную микрофлору макроорганизма, не подавляют его механизмы иммунной защиты и не обладают токсическим действием. На литическую активность бактериофагов не влияет наличие резистентности бактерий к антибиотикам. Главным условием клинической эффективности при назначении препаратов бактериофагов является фагочувствительность бактериального возбудителя.
Организация производства лечебно-профилактических препаратов бактериофагов, как и других иммунобиологических лекарственных средств, осуществляется согласно санитарным правилам Союза и государств-членов, с учетом требований системы обеспечения качества надлежащей производственной практики Союза. Вместе с тем имеются определенные особенности в комплектации и размещении производственных помещений.
Для работы со штаммами бактерий и фагами, выделенными из патологического материала и объектов внешней среды (почва, водоемы, сточные воды и т.д.) необходимы изолированные от производства боксированные помещения для проведения микробиологических исследований.
Обязательными начальными этапами работы являются:
выделение чистых культур перспективных штаммов бактерий (т.е. кандидатов в производственные штаммы бактерий);
выделение вирулентных бактериофагов с широким спектром антибактериальной активности для пополнения коллекции маточных фагов.
В производственной зоне должны быть предусмотрены раздельные микробиологические боксы для работы с производственными бактериальными штаммами и маточными фагами.
При производстве фаговых препаратов проводят валидацию технологического процесса, технологического оборудования, сырья и методов контроля. Все исходные материалы и сырье, используемые при производстве, должны иметь документы, подтверждающие их качество. Вспомогательные вещества, входящие в состав препаратов с лечебно-профилактическими бактериофагами, должны быть разрешены к медицинскому применению и использоваться в дозах, не вызывающих токсические, аллергические или иные нежелательные реакции у человека.
Производственные питательные среды не должны содержать антибиотиков и компонентов, вызывающих аллергические или иные нежелательные реакции у человека. Питательные среды должны обладать хорошими ростовыми свойствами и быть стерильными. Сырье, реактивы и реагенты, используемые при производстве питательных сред, должны быть пригодны для соответствующих целей и их качество должно быть подтверждено документально.
1.2. Требования к производственным штаммам
Производственные штаммы бактерий - продуценты бактериофагов, которые выделяют от больных гнойно-септическими или кишечными инфекциями и получают из бактериологических диагностических лабораторий, расположенных в регионах реализации и потребления лечебно-профилактических бактериофагов. Коллекция производственных штаммов бактерий, используемых в производстве бактериофагов, должна ежегодно обновляться свежевыделенными штаммами от больных не менее чем на одну треть.
Производственные штаммы бактерий должны обладать типичными для каждого вида морфологическими, культуральными, биохимическими и серологическими свойствами, не продуцировать энтеротоксины и не содержать умеренных бактериофагов. Оценку наличия индуцируемых профагов проводят путем обработки бактерий индукторами (ультрафиолетовым излучением, митомицином C, налидиксовой кислотой) с последующим контролем появления бляшек лизиса, вызванного активацией умеренных бактериофагов, содержащихся в геноме бактерии.
Производственные штаммы бактерий должны лизироваться маточными фагами в титрах по методу Аппельмана не менее чем на 1 - 2 порядка выше показателей специфической активности конечного продукта. При этом стабильность лизиса должна сохраняться после 48 часов инкубирования при температуре 37 °C. Температурный режим и время инкубации зависят от специфических особенностей бактерий и их фагов.
Производственные штаммы бактерий хранят в специальных изолированных помещениях в лиофилизированном состоянии в ампулах при температуре 2 - 8 °C в течение 10 лет или в бактериологических пробирках с 0,4 - 0,7 процентной агаризованной питательной средой (на основе гидролизата Хоттингера, МПБ или других питательных сред в зависимости от специфических потребностей бактерий) под стерильным вазелиновым маслом, при температуре 2 - 8 °C и регулярном пересеве каждые 2 - 3 месяца.
В качестве контрольных бактериальных штаммов не должны использоваться производственные штаммы бактерий, используемые для наработки соответствующих бактериофагов.
Маточные бактериофаги.
Для получения фаговых препаратов используются только вирулентные бактериофаги. Их выделяют из природных источников - клинического материала, сточных вод, почвы, пассируя на штаммах целевых видов бактерий - свежевыделенных и производственных. Подбирают высокоактивные расы (штаммы) фагов к слаболизирующимся и фагорезистентным штаммам бактерий. Пополнение производственных фаговых рас различными штаммами из природных источников позволяет преодолевать первичную фагоустойчивость возбудителей. Маточные бактериофаги должны включать вирулентные фаги с широким диапазоном действия по отношению к штаммам соответствующего вида бактерий, обладать высокой активностью, стабильностью лизиса, специфической направленностью антимикробного действия и высокой урожайностью. Характеристика кандидатных фаговых рас (штаммов) при отборе может быть дополнена электронно-микроскопическим изучением их морфологии и другими современными молекулярно-биологическими методами (полногеномное секвенирование с последующим биоинформационным анализом). Хранение производственной коллекции бактериофагов осуществляется в жидком состоянии при температуре от 2 до 8 °C в течение 5 лет с ежегодным пересевом на бактериальных штаммах или в лиофилизированном состоянии.
Расширение диапазона действия препаратов в отношении циркулирующих бактериальных возбудителей может быть достигнуто путем подбора фаговых рас из коллекции предприятия или из природных источников. Это обеспечивает возможность выпуска производственных серий бактериофагов целевого назначения. Например, при вспышке или появлении очага инфекции, обусловленной фагоустойчивым возбудителем, передача на производство эпидемически значимого бактериального штамма позволит адаптировать препарат к конкретным эпидемиологическим условиям путем подбора к этому бактериальному штамму активных фаговых рас из коллекции производителя и последующего использования их при изготовлении препарата.
1.3. Основные этапы производства
Производственный процесс включает в себя: работу с бактериальными возбудителями гнойно-септических и кишечных инфекций, подбор к ним вирулентных штаммов бактериофагов, отбор бактерий - продуцентов бактериофагов, получение маточных бактериофагов, культивирование производственных штаммов бактерий - продуцентов и маточных бактериофагов с целью получения биомассы бактериофагов и приготовление препаратов бактериофагов. Освобождение фаголизитов от продуктов жизнедеятельности бактерий, эндо- и экзотоксинов, продуктов фаголизиса бактериальных клеток должно быть обеспечено дополнительными методами очистки (ультрафильтрацией, афинной хроматографией и т.д.).
После завершения процесса фаголизиса бактериальной взвеси осуществляется стерилизующая фильтрация фаголизатов, затем очищение от бактериальных метаболитов и токсинов, компонентов питательной среды, далее фаголизаты подвергаются концентрированию, полученные концентраты стерилизуются.
После концентрирования из фаголизатов готовят:
жидкие препараты, которые доводятся до финального титра буферными растворами pH 6,0 - 8,0, стерилизуют и разливают в стерильные флаконы, укупориваются герметично и стерильно;
лиофилизированную биомассу, используемую для получения таблеток, суппозиториев, мазей, линиментов и др.
1.4. Контроль качества в процессе производства
В процессе производства бактериофагов на разных этапах получают промежуточные продукты, которые должны соответствовать определенному уровню качества.
При подготовке производственных штаммов бактерий контроль посевных культур бактериальных штаммов-продуцентов включает в себя следующие показатели:
чистота;
типичность биологических свойств;
отсутствие легко индуцируемых профагов.
При подготовке маточных бактериофагов контроль маточных бактериофагов включает в себя следующие показатели:
содержание фаговых частиц в 1 мл, определяемое методом агаровых слоев по Грациа;
специфическая активность, определяемая титрованием в жидкой питательной среде по методу Аппельмана на посевных бактериальных штаммах;
стабильность лизиса (сохранность полученных результатов лизиса по методу Аппельмана) в течение 48 часов инкубации при температуре 37  1 °C. Температурный режим и время инкубации зависят от специфических особенностей бактерий и их фагов;
1 °C. Температурный режим и время инкубации зависят от специфических особенностей бактерий и их фагов;
стерильность.
По мере завершения очистки фаголизатов, концентрирования, стерилизующей фильтрации проводится определение показателей pH, стерильности, специфической активности по методу Аппельмана.
Для получения жидкого препарата после финальной стадии бактериофаг разливают по флаконам и укупоривают.
Для получения других лекарственных форм препаратов бактериофагов:
в концентрированный бактериофаг добавляют стабилизаторы и лиофилизируют. Сухую массу бактериофага контролируют в тестах на специфическую активность и микробиологическую чистоту;
после добавления наполнителей осуществляют технологические процессы, соответствующие заданной лекарственной форме (таблетки, капсулы, суппозитории, линименты, мази);
готовый продукт контролируют по всем показателям, соответствующим лекарственной форме.
К специфическим методам исследования, используемым для характеристики лечебно-профилактических бактериофагов, относятся методы Аппельмана и Грациа.
1.5. Транспортирование и хранение
Транспортирование осуществляется при температуре от 2 до 8 °C, в пределах 1 месяца - допускается при температуре от 9 до 25 °C.
Хранение в сухом, защищенном от света и недоступном для детей месте при температуре от 2 до 8 °C в течение 1 - 2 лет. Однако возможно расширение температурного режима хранения до 25 °C при подтверждении активности препарата в течение заявленного срока годности.
2. Доклинические исследования
лечебно-профилактических бактериофагов
Оценка возможности использования новых фаговых препаратов в лечебных целях должна проводиться с учетом современных знаний о биологии, генетике и экологии бактериальных вирусов. В отчетах по доклиническим испытаниям лечебно-профилактических препаратов бактериофагов должны быть представлены результаты изучения биологических свойств препарата (в том числе биологических свойств фаговых штаммов, включенных в его состав), результаты изучения эффективности фагового препарата при лечении экспериментальной бактериальной инфекции, безопасности (острой и хронической токсичности, аллергизирующих свойств).
2.1. Биологические свойства
лечебно-профилактических бактериофагов
При конструировании препаратов используют вирулентные фаги природного происхождения. Исходной субстанцией лечебно-профилактических бактериофагов являются стерильные фильтраты фаголизатов соответствующих видов бактерий. Дополнительными методами очистки должно быть обеспечено их освобождение от продуктов жизнедеятельности бактерий, эндо- и экзотоксинов, продуктов фаголизиса бактериальных клеток. Очищенные концентрированные фаголизаты подлежат соответствующим технологическим процедурам в зависимости от вида конечного препарата (моно- или комбинированный) и его лекарственной формы. В состав препарата должны входить несколько штаммов (не менее 2 - 3) вирулентных фагов, биологические свойства каждого из них должны быть охарактеризованы.
2.1.1. Характеристика биологических свойств фаговых штаммов, включенных в состав лечебно-профилактического препарата бактериофагов.
Бактериофаги должны иметь строго литический инфекционный цикл. Наиболее полной характеристикой, позволяющей всесторонне оценить наличие генов перехода в лизогенный цикл, факторов вирулентности и токсинов, является определение полной последовательности генома бактериофага.
Экспериментальными признаками такой классификации могут служить определение морфологии вирусной частицы с помощью электронной микроскопии негативного контрастирования и ПЦР-типирование по наиболее консервативным генам, характерным для данной группы бактериофагов.
Каждый из фаговых штаммов, используемых в препарате, должен:
демонстрировать высокую специфическую литическую активность, выявляемую путем титрования в жидкой питательной среде по методу Аппельмана;
иметь высокую концентрацию (количество бляшкообразующих единиц в 1 мл), определяемую методом агаровых слоев по Грациа;
обладать широким диапазоном литического действия на представительной коллекции бактериальных штаммов, музейных и изолированных от больных;
быть изучен с помощью молекулярно-генетических методов (полногеномного секвенирования) для определения его полной нуклеотидной последовательности ДНК с последующим биоинформационным анализом;
быть морфологически охарактеризован с помощью электронной микроскопии негативного контрастирования, что позволяет соотнести de novo выделенный бактериофаг с известными группами фагов, которые заведомо относятся к литическим и пригодным для терапевтического применения.
Другими характеристиками, которые должны быть определены для каждого фагового штамма, являются:
частота генерации фагоустойчивых форм;
длительность латентного периода в ходе инфекции;
урожайность и величина выхода фаговых частиц из одной инфицированной бактериальной клетки.
Кроме того, должна быть представлена характеристика бактериального штамма, на котором проводится культивирование бактериофага. Штамм не должен содержать легко индуцируемых профагов, загрязняющих препарат. Оценку наличия индуцируемых профагов проводят путем обработки бактерий индукторами (ультрафиолетовым излучением, митомицином C, налидиксовой кислотой) с последующим контролем появления бляшек лизиса, вызванного активацией умеренных бактериофагов, содержащихся в геноме бактерии.
2.1.2. Характеристика основных биологических свойств лечебно-профилактических препаратов бактериофагов.
Представленные материалы на препарат в целом, помимо характеристик на отдельные фаги, должны содержать следующую информацию:
специфическая литическая активность, определяемая по методу Аппельмана с использованием соответствующих бактериальных тест-штаммов (контрольных штаммов). Тест-штаммы (не менее 10), типичные по своим морфологическим, культуральным, биохимическим свойствам, не должны быть использованы при изготовлении препарата. Активность бактериофага, определяемая по методу Аппельмана, выражается отрицательным десятичным логарифмом, указывающим последнее разведение бактериофага в жидкой питательной среде, в которой рост бактериальной культуры визуально не наблюдается;
диапазон действия изучаемого препарата в отношении соответствующих ему видов бактериальных возбудителей инфекций, выделенных от больных (не менее 70 процентов фагочувствительных штаммов), циркулирующих в современных условиях на территориях государств-членов (не менее 3-х регионов);
стабильность препарата. Активность препаратов бактериофагов должна сохраняться в течение всего заявленного срока годности при регламентированной температуре хранения. Определение стабильности препарата (способности сохранять литическую активность бактериофага) проводится по методу Аппельмана.
При проведении исследования антимикробного действия бактериофагов необходимо обеспечение следующих условий, гарантирующих достоверность результатов:
питательные среды для культивирования бактерий, на которых изучается антимикробное действие бактериофага, должны отвечать требованиям стандартности и воспроизводимости результатов;
стандартность бактериальной нагрузки (посевной дозы) штаммов-мишеней для изучаемого бактериофага.
2.2. Определение эффективности
Эффективность фаговой терапии должна быть установлена на соответствующей модели экспериментальной инфекции. Изучение защитного действия фагового препарата проводится в отношении бактериального возбудителя, против которого разработан изучаемый препарат.
Экспериментальные животные.
При изучении терапевтической эффективности используются различные виды животных: белые мыши и крысы, морские свинки, кролики. На первых этапах целесообразно использование белых мышей. В исследованиях in vivo на животных подтверждается антибактериальный эффект, выявленный в исследованиях in vitro.
Модели экспериментальной инфекции.
Наиболее распространенной моделью является генерализованная инфекция (экспериментальный сепсис) белых мышей (массой 18 - 20 г, не менее 10 особей в каждой группе). Заражающая доза для каждого вида бактерий устанавливается в предварительных опытах.
Способы заражения - внутривенное, внутрибрюшинное, подкожное или внутримышечное (последние 2 варианта - при использовании высоковирулентных возбудителей).
Выбор других моделей экспериментальной инфекции зависит от специфики возбудителя, его тропности к тем или иным системам организма. Возможно моделирование хронической септикопиемии, пневмонии, пиелонефрита, менингита и менингоэнцефалита, других локализованных инфекционных процессов.
Порядок проведения исследования.
После выявления эффективности препарата in vivo на этапе первичного скрининга проводятся исследования терапевтической эффективности по сравнению с антибактериальными препаратами (антибиотиками) той же направленности, что и изучаемый препарат. При оптимальной дозе введения определяется связь эффективности препарата с тяжестью инфекции (при различных инфицирующих дозах - 1 LD50, 2 - 4 LD50, 10 LD50, 100 LD50 и более в зависимости от возбудителя).
В зависимости от цели исследования проводится оценка:
профилактической эффективности препарата - введение препарата за 1, 3, 6, 12, 24 и 48 часов до заражения;
терапевтической эффективности препарата - введение препарата через 1, 3, 6, 12, 24 или 48 часов после заражения.
Исследования проводятся при различной кратности введения (однократно или повторно в зависимости от модели). Традиционная модель фаготерапии - внутрибрюшинное введение препарата с определенным содержанием фаговых частиц в вводимой дозе ежедневно в течение 10 - 12 суток. В процессе исследования определяют минимальную эффективную дозу с учетом предполагаемой терапевтической дозы для человека в перерасчете на килограмм массы.
Каждое отдельное исследование включает в себя 2 вида контроля:
контроль гибели животных без лечения;
контроль эффективности лечения (в сравнении с известным антимикробным препаратом - антибиотиком аналогичной направленности).
В качестве препарата сравнения можно использовать питательную среду, на которой приготовлен изучаемый препарат бактериофага.
Длительность наблюдения за животными зависит от особенности течения инфекции при конкретной нозологической форме.
Оценка эффективности.
Оценка эффективности препарата бактериофага проводится по следующим показателям:
сравнительная выживаемость и сроки гибели животных в контрольной и опытной группах;
динамика освобождения от возбудителя в контрольной и опытной группах (в процессе лечения и по его окончании проводят посев крови, органов с определением числа бактерий на 1 г);
клинические проявления (температура тела, изменение веса, состояние шерстяного покрова, поведенческие реакции);
клинико-лабораторные показатели (клинический анализ крови, показатели анализа спинномозговой жидкости и др.);
патоморфологические изменения органов и тканей в процессе лечения и по его окончании.
Результаты обрабатываются адекватными статистическими методами.
2.3. Безопасность
Препараты бактериофагов должны отвечать всем требованиям безопасности и не иметь бактериальных или иных токсических загрязнений. Современные технологии изготовления фаговых препаратов предусматривают высокую степень очистки от продуктов жизнедеятельности бактерий, эндо- и экзотоксинов, продуктов фаголизиса бактериальных клеток. Подтверждением этому могут быть исследования препаратов по определению бактериальных эндотоксинов, энтеротоксинов и др. в соответствии с особенностями изучаемого препарата.
Методы очистки препаратов должны быть подробно описаны, обоснованы и включены в отчеты по результатам доклинических исследований.
Подтверждение безопасности препаратов бактериофагов включает в себя изучение токсичности (острой и хронической), местного и аллергизирующего действия.
Исследование токсичности.
Определение токсичности фаговых препаратов проводят на белых мышах массой 18 - 20 г, каждая группа должна включать не менее 10 животных. Необходимость проведения гистологических исследований зависит от способности бактериальных штаммов, используемых при производстве бактериофагов, к токсинообразованию.
Оценка токсичности при однократном введении.
Исследование острой токсичности проводится при однократном внутрибрюшинном введении фага в дозе, многократно превышающей максимальную разовую дозу для человека в пересчете на единицу массы тела (100 - 200 раз). Максимальный объем - не более 1 мл. Срок наблюдения - не менее 7 суток после введения препарата. Животные не должны терять в весе, должны отсутствовать признаки заболевания и инфильтраты в месте введения препарата. В периферической крови опытных животных после введения бактериофага должны отсутствовать значительные изменения картины крови по сравнению с данными в контрольной группе. Необходимо проведение гистологических исследований внутренних органов животных.
Оценка токсичности при многократном введении.
Изучение токсичности при многократном введении проводится при внутрибрюшинном введении фага ежедневно в течение 20 дней в дозе, максимально эквивалентной разовой дозе для человека в пересчете на единицу массы тела.
Наблюдения за животными проводят в течение всего периода введения и последующих 7 суток с ежедневной регистрацией массы тела животных, а также возможных клинических симптомов интоксикации. При исследовании периферической крови опытных животных после введения бактериофага не должны обнаруживаться значительные изменения в содержании лейкоцитов по сравнению с контрольной группой. Необходимо проведение гистологических исследований внутренних органов животных (не менее чем 5 особей) сразу после курса фаготерапии и после 7-дневного восстановительного периода
Определение местного действия бактериофага осуществляется в случае использования препаратов внутримышечно в дозе, предлагаемой для применения. Оценку проводят на основании данных осмотра на протяжении всего срока введения препарата и последующих 7 суток.
Оценка аллергизирующих свойств.
Большинство бактериофагов при первичном применении являются нео-антигенами для организма животных и человека, т.е. заранее имеющихся против них антител в организме нет. Экспериментально показано, что при однократном приеме фильтрата фаголизата иммунный ответ на присутствие фагов в организме незначителен и не влияет на терапевтический эффект фага. Отечественными исследователями доказано, что введение очищенных фаговых препаратов в терапевтических дозах в течение 15 - 20 дней не стимулирует образования антифаговых антител. Вместе с тем имеются данные, что при более длительном применении фага стимуляция адаптивной иммунной системы потенциально вероятна.
Изучение аллергизирующих свойств новых препаратов бактериофагов проводится в соответствии с традиционными методами исследования препаратов МИБП по определению ГНТ и ГЗТ.
Фармакокинетика фаговых препаратов.
Бактериофаги не подвергаются какому-либо специфическому метаболизму в макроорганизме. Результаты изучения особенностей циркуляции фаговых частиц при моделировании инфекционного процесса могут использоваться в качестве дополнительных данных по исследованию нового препарата.
3. Клинические исследования
лечебно-профилактических бактериофагов
Поскольку фаговые препараты обладают строгой специфичностью действия, клинические испытания должны предусматривать только этиотропную антибактериальную терапию или профилактирование развития инфекций, обусловленных видами бактерий, соответствующими изучаемому препарату. В этой связи клиническим наблюдениям обязательно предшествует работа по выявлению и сбору бактериальных возбудителей и определению их чувствительности к изучаемому фаговому препарату. На основании полученных результатов может быть расширен диапазон действия препарата за счет его адаптации к устойчивым штаммам.
При организации клинических испытаний необходимо учитывать фагочувствительность штаммов, являющихся возбудителями инфекции, подлежащей лечению. При этом должна быть обеспечена полноценная материальная база для микробиологических исследований.
В настоящее время наибольшую социальную значимость приобрели гнойно-септические заболевания, связанные с пребыванием в стационарах. Все больше видов условно-патогенных бактерий становятся патогенными для ослабленных больных в госпитальных условиях. Для этих штаммов характерны множественная устойчивость к антибиотикам и нарастание вирулентности. Поэтому выбор клинической базы должен основываться на анализе этиологической структуры бактериальных инфекций. Эпидемиологически значимые штаммы, изолированные от больных, полученные из смывов с медицинского оборудования или с объектов больничной среды, подлежат изучению на чувствительность к исследуемому фаговому препарату. Оптимальной моделью для клинических испытаний лечебно-профилактических бактериофагов являются госпитальные инфекции. Изучаемый препарат бактериофагов должен лизировать возбудителей целевых инфекций.
Целью клинических исследований новых лечебно-профилактических препаратов бактериофагов является изучение их эффективности при различных нозологических формах бактериальных инфекций, а также их безопасность и переносимость пациентами с инфекцией различной локализации и степени тяжести. В этой связи подлежат решению следующие задачи:
определение клинической эффективности фагового препарата;
определение микробиологической эффективности фагового препарата;
определение безопасности и переносимости фагового препарата пациентами с различными проявлениями инфекции соответствующей этиологии при различной локализации и степени тяжести;
отработка наиболее оптимальных доз, рациональных схем и длительности применения нового фагового препарата при различных нозоформах и степени тяжести изучаемой инфекции;
сопоставление клинической и микробиологической эффективности и переносимости фагового препарата с антимикробными препаратами, применяемыми при терапии различных нозологических форм данной инфекции.
4. Отработка показаний к назначению исследуемого препарата
и рекомендаций по его использованию в медицинской практике
В качестве групп сравнения должны вестись наблюдения за больными с аналогичными заболеваниями соответствующей этиологии, нозоформами и степенью тяжести, получающими традиционную антимикробную терапию.
Дизайн исследования (методология).
Основанием для начала проведения клинических исследований являются результаты отчета по доклиническому изучению нового фагового препарата, проведенному in vitro и на животных.
При планировании и составлении протокола клинических исследований необходимы четкое описание методов выделения и идентификации возбудителей инфекции, оценка клинической значимости выделенных микроорганизмов, их количественная оценка с критериями интерпретации по результатам терапии, описание методов определения чувствительности бактерий к бактериофагам.
При планировании клинических исследований нового фагового препарата необходимо предусмотреть следующее:
основные (первичные) и второстепенные (вторичные) критерии клинической эффективности при каждой нозологической форме наблюдаемой инфекции;
вид исследования (проспективное, двойное слепое, рандомизированное исследование в параллельных группах);
рандомизация пациентов с указанием параметров, например, по степени тяжести, длительности заболевания, предшествующей антимикробной терапии;
дозировка и режим введения, продолжительность курса;
возможность сочетанного применения с базисной антибиотикотерапией, в первую очередь при выявлении нескольких видов бактериальных возбудителей (микст-инфекция);
условия отмены исследуемого препарата или прерывания клинических исследований.
Критерии включения пациентов в клинические исследования.
Лечебно-профилактические бактериофаги являются антибактериальными препаратами узкоспецифической направленности, подавляющими только целевых (гомологичных) возбудителей. Обязательным условием проведения испытаний является фагочувствительность бактериальных штаммов, вызвавших развитие инфекции.
Поскольку антибактериальное действие бактериофагов в отличие от антибиотиков развивается постепенно, для испытания фаговых препаратов в виде монотерапии целесообразно включение больных с нетяжелыми проявлениями изучаемой инфекции. При определенных условиях (по клиническим показаниям) у части наблюдаемых возможно совместное назначение бактериофагов и антибиотиков (в качестве базисной антибиотикотерапии). При этом аналогичная базисная антибиотикотерапия должна проводиться в группе сравнения.
В клинические исследования включаются больные:
у которых выявленный возбудитель чувствителен к изучаемому препарату;
не получавшие антимикробные препараты по поводу данного заболевания или сопутствующих заболеваний;
начавшие получать антимикробную терапию (не более 1 дозы) до включения в исследование, а также через 2 - 3 дня после включения (кроме антибиотиков с коротким курсом - менее 3 суток).
Критериями невключения для клинических исследований новых фаговых препаратов являются анамнестические данные о серьезных неблагоприятных лекарственных реакциях, в том числе о каких-либо аллергических проявлениях, а также о тяжелых сопутствующих соматических заболеваниях. Беременность и лактация не являются противопоказаниями, поскольку бактериофаги являются вирусами бактерий и не обладают тропизмом к клеткам других организмов. Накопленный в течение длительного периода успешный опыт использования лечебно-профилактических бактериофагов при лечении гнойно-воспалительных заболеваний у беременных и кормящих (лактирующих) женщин свидетельствует о безвредности этих препаратов для данной категории пациентов.
Первую фазу клинических исследований проводят для определения реактогенности и безопасности нового лечебно-профилактического фагового препарата для человека, как правило, на здоровых добровольцах, однако может быть получено разрешение на ограниченные исследования у пациентов с нетяжелыми проявлениями инфекции, вызванной бактериальным возбудителем, гомологичным фаговому препарату.
В ходе клинических испытаний первой фазы должны контролироваться клинические и лабораторные данные для выявления нежелательных реакций. В число обследований, как правило, должны входить развернутый клинический анализ крови и клинический анализ мочи. Кроме того, необходима микробиологическая диагностика инфекционного заболевания с количественной характеристикой и определением фагочувствительности возбудителя, предваряющая назначение препарата. Включение пациента в группу наблюдения возможно лишь при выявлении чувствительности бактериального штамма к изучаемому препарату.
После завершения клинических испытаний первой фазы и подтверждения безопасности нового фагового препарата следует продолжение клинических испытаний второй фазы.
Вторую фазу клинических испытаний проводят в виде контролируемых наблюдений за больными с нетяжелыми проявлениями инфекции, вызванной бактериальным возбудителем, чувствительным к изучаемому фаговому препарату. При формировании групп следует учитывать, что у пациентов не должно быть серьезных сопутствующих заболеваний. Изучение эффективности препарата проводят в сопоставлении со стандартными антибактериальными препаратами для лечения данной инфекции. В качестве препаратов сравнения используют антибиотики.
В ходе проведения клинических испытаний второй фазы отрабатываются дозировка и продолжительность курса в зависимости от формы или степени тяжести заболевания. При этом должны контролироваться клинические и лабораторные данные, в том числе развернутый клинический анализ крови и клинический анализ мочи. Кроме того, необходима микробиологическая диагностика инфекционного заболевания с количественной характеристикой и определением чувствительности возбудителя к исследуемому фаговому препарату и антибиотикам. Эти исследования проводят перед началом и по окончании наблюдения.
Проводится анализ состояния пациентов в течение 3 - 4 недель после завершения курса фаготерапии и базисной антимикробной терапии в основной и контрольной группах для определения окончательного (отдаленного) клинического и микробиологического исхода лечения и выявления отдаленных нежелательных реакций. Следует обратить особое внимание на возможность формирования устойчивости возбудителей к антимикробным препаратам (в первую очередь к антибиотикам).
После успешно прошедших клинических испытаний второй фазы проводится третья фаза клинических испытаний в виде расширенных контролируемых наблюдений, главной целью которых служит оценка эффективности и безопасности нового фагового препарата в условиях, максимально приближенных к клинической практике.
Наблюдению подлежат большие группы пациентов (от нескольких сот человек). В протоколе необходимо указать, какие действия следует предпринять в случае неэффективности фаготерапии (определенные диагностические исследования, коррекцию антибактериальной терапии или раскрытие кода рандомизации, если это необходимо в сложившейся клинической ситуации).
Четвертую фазу клинических испытаний проводят после регистрации препарата. По своей организации они подобны клиническим исследованиям третьей фазы и проводятся для оценки клинической эффективности препарата или изучения отдельных аспектов (например, фармакоэкономические преимущества определенных аспектов фаготерапии) на большой и разнородной популяции пациентов.
Все клинические исследования, в том числе третьей и четвертой фазы, предполагают наблюдения за больными с различными проявлениями инфекции, вызванной бактериальным возбудителем, чувствительным к изучаемому фаговому препарату.
Критерии определения эффективности лечебно-профилактических бактериофагов
При клинических испытаниях оценивается как лечебная, так и микробиологическая эффективность препарата, поэтому в протоколе необходимо подробно изложить принципы оценки результатов.
Лечебная эффективность предполагает 3 категории:
эффективность - полное исчезновение всех субъективных и объективных клинических признаков заболевания, уменьшение клинических проявлений или отсутствие прогрессирования по данным объективных исследований;
неэффективность - ухудшение состояния, нарастание всех субъективных и объективных клинических признаков заболевания, появление новых симптомов, летальный исход;
эффективность невозможно оценить - выход пациентов из исследования по каким-либо причинам, в том числе летальный исход во время проведения исследований, не связанный с данным заболеванием.
Обязательно используются показатели лабораторных исследований.
Определение микробиологической эффективности.
Микробиологическая эффективность оценивается после завершения курса фаготерапии и через 3 - 4 недели после проведения курса лечения при последующем наблюдении за пациентом. Возможны следующие оценки результатов терапии:
элиминация возбудителя - прекращение высева (выявления) возбудителя из очага первичной локализации инфекции;
снижение уровня присутствия возбудителя в биологическом материале из очага первичной локализации инфекции. Значительное снижение - на 2 - 3 порядка и более - свидетельствует о наличии микробиологической эффективности;
смена возбудителя или суперинфекция;
колонизация - обнаружение новых микроорганизмов, отличающихся от первоначального возбудителя, в местах первичной локализации инфекции или в других биотопах при отсутствии признаков активного инфекционного процесса.
При наличии микст-инфекции, вызванной двумя или несколькими бактериальными агентами, микробиологическая эффективность оценивается по каждому виду бактериального агента отдельно.
По остальным позициям (оценка безопасности, нежелательные реакции, методы анализа и представления данных) анализ материала проводится согласно требованиям правил надлежащей клинической практики Союза.