РЕКОМЕНДАЦИИ
ПО ОСУЩЕСТВЛЕНИЮ АДМИНИСТРАТИВНЫХ ПРОЦЕДУР И ДЕЙСТВИЙ
В РАМКАХ ПРЕДОСТАВЛЕНИЯ ГОСУДАРСТВЕННОЙ УСЛУГИ ПО ПРОДЛЕНИЮ
СРОКА ДЕЙСТВИЯ ИСКЛЮЧИТЕЛЬНОГО ПРАВА НА ИЗОБРЕТЕНИЕ
И УДОСТОВЕРЯЮЩЕГО ЭТО ПРАВО ПАТЕНТА
Используемые сокращения
Кодекс - Гражданский кодекс Российской Федерации.
Регламент - Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патента, утвержденный приказом Министерства экономического развития России от 3 ноября 2015 г. N 810, зарегистрированный в Министерстве юстиции Российской Федерации 25 декабря 2015 г. N 40250, с изменениями.
Порядок - Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, утвержденный приказом Министерства экономического развития России от 3 ноября 2015 г. N 809, зарегистрированный в Министерстве юстиции Российской Федерации 25 декабря 2015 г. N 40249.
Положение о пошлинах - Положение о патентных и иных пошлинах за совершение юридически значимых действий, связанных с патентом на изобретение, полезную модель, промышленный образец, с государственной регистрацией товарного знака и знака обслуживания, с государственной регистрацией и предоставлением исключительного права на наименование места происхождения товара, а также с государственной регистрацией отчуждения исключительного права на результат интеллектуальной деятельности или средство индивидуализации, залога исключительного права, предоставления права использования такого результата или такого средства по договору, перехода исключительного права на такой результат или такое средство без договора, утвержденное постановлением Правительства Российской Федерации от 10.12.2008 N 941 с изменениями, внесенными постановлением Правительства Российской Федерации от 15.09.2011 N 781, с изменениями, внесенными постановлением Правительства Российской Федерации от 14.11.2013 N 1023, от 22.03.2016 N 227 (Собрание законодательства Российской Федерации, 2008, N 51, ст. 6170; 2011 N 39, ст. 5487; 2013 N 47, ст. 6106; 2016, N 13, ст. 1844, 2017, N 40, ст. 5856 официальный интернет-портал правовой информации www.pravo.gov.ru, 28.09.2017).
Рекомендации - рекомендации по осуществлению административных процедур и действий в рамках предоставления государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патента.
Роспатент - Федеральный орган исполнительной власти по интеллектуальной собственности.
Заявитель - правообладатель патента на изобретение.
Заявление - заявление о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента.
Документы - Заявление и прилагаемые к нему необходимые документы.
Государственный реестр - Государственный реестр изобретений Российской Федерации.
ЕПГУ - Единый портал государственных и муниципальных услуг.
ГИС ГМП - Государственная информационная система о государственных и муниципальных платежах.
1. Введение
Настоящие Рекомендации по осуществлению административных процедур и действий в рамках предоставления государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патента разработаны с целью разъяснения положений Регламента, разъяснения отдельных ситуаций, возникающих при подаче и рассмотрении Заявления.
Рекомендации направлены на обеспечение реализации положений Гражданского кодекса Российской Федерации, касающихся продления срока действия исключительного права на изобретение и удостоверяющего это право патента, которое подлежит подаче в Федеральный орган исполнительной власти по интеллектуальной собственности (далее - Роспатент).
В Рекомендациях рассматриваются вопросы, связанные с проверкой документов на соответствие требованиям, необходимым для удовлетворения заявления.
Рекомендации разработаны для экспертов и специалистов Роспатента и подведомственных ему организаций с целью их применения при проверке документов, касающихся продления срока действия исключительного права на изобретение и удостоверяющего это права патента.
Предлагаемые в Рекомендациях методические подходы, определены исходя из положений Гражданского кодекса Российской Федерации и Административного регламента предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патента.
2. Общие положения
Продление срока действия исключительного права на изобретение, относящееся к лекарственному средству, пестициду и агрохимикату, впервые в отечественной практике было введено в Патентный закон Российской Федерации в 2003 г. Федеральным законом от 07.02.2003 N 22-ФЗ "О внесении изменений и дополнений в Патентный закон Российской Федерации". Затем эта норма без изменений была перенесена в часть четвертую Гражданского Кодекса Российской Федерации (далее - Кодекс) и продолжила свое действие до вступления в силу Федерального закона от 12.03.2014 N 35. Этим законом были внесены изменения в Кодекс, в том числе в норму, касающуюся порядка продления срока действия патента на изобретение.
Административно-процедурное регулирование вопросов продления срока действия исключительного права на изобретение с 08.01.2016 осуществляется на основании пакета нормативных установлений Регламента и Порядка.
2.1. Изменения, введенные новыми нормативно-правовыми актами
В соответствии с пунктом 2 статьи 1363 Кодекса с 01.01.2015 продление срока действия исключительного права осуществляется путем выдачи дополнительного патента с новой формулой изобретения, содержащей совокупность признаков запатентованного изобретения, характеризующей продукт, на применение которого получено разрешение.
Это означает, что если раньше продлевался срок действия патента в отношении независимых и зависимых пунктов формулы изобретения, с которыми этот патент был выдан, то с 2015 г. действие исключительного права продлевается только на конкретное соединение или композицию лекарственного средства, которое проходило клинические испытания и на применение которого было получено разрешение.
При продлении срока действия исключительного права на изобретение предусматривается выдача дополнительного патента, который будет иметь регистрационный номер, соответствующий номеру первоначально выданного патента на изобретение, внесенное в Государственный реестр изобретений Российской Федерации.
Заявление о продлении срока действия исключительного права на изобретение, предполагающее выдачу дополнительного патента при условии соблюдения соответствующих условий, должно быть подано патентообладателем в период действия основного патента до истечения шести месяцев со дня получения первого разрешения на применение "данного продукта" или до истечения шести месяцев с даты выдачи основного патента (в зависимости от того, какой срок истекает позднее).
Срок действия исключительного права продлевается на время, прошедшее с даты подачи заявки на изобретение до дня получения первого разрешения на применение продукта, за вычетом пяти лет, но не более чем на пять лет.
Одно из существенных изменений этой нормы заключается в том, что дополнительный патент будет удостоверять новый объем прав, поскольку будет содержать новую формулу изобретения, которая будет характеризовать конкретный продукт, на применение которого получено первое разрешение.
Продукт, на применение которого получено разрешение, - это активное соединение или комбинация активных соединений, а также их соли, которые проходили клинические испытания, и на применение которых в качестве лекарственного средства получено Регистрационное удостоверение Минздрава.
Под продуктом также следует понимать фармацевтическую композицию, в виде различных лекарственных форм, на применение которых также было получено Регистрационное удостоверение Минздрава.
Для сравнения необходимо отметить, что если ранее исключительное право продлевалось, например, на всю формулу Маркуша, являющуюся "зонтиком", который накрывает собой бесчисленное множество альтернативных соединений, то принятые изменения позволят продлить исключительное право только лишь на конкретное активное соединение, которое проходило испытания и на применение которого получено Регистрационное удостоверение.
2.2. Новый алгоритм рассмотрения заявления о продлении
Наряду с решением формально-административных вопросов проверки уплаты соответствующей патентной пошлины, соблюдения срока представления заявления о продлении исключительного права, проверки статуса основного патента и правомочий лица, подавшего заявление, в первую очередь будет устанавливаться возможность отнесения изобретения к той категории изобретений, в отношении которой предусматривается возможность продления срока действия патента.
Положительное решение будет принято только в том случае, если в формуле изобретения продукт будет охарактеризован в виде соединения или группы соединений, предназначенных для изготовления лекарственных средств, а также продукт будет представлять собой композицию лекарственного средства.
Продление срока действия патента на изобретение, относящееся к устройству или способу, в котором лекарственное средство применяется, не допускается.
Далее, при анализе возможности выдачи дополнительного патента будет проводиться проверка идентичности совокупности признаков, определяющих объем правовой охраны, характеризующих соединение или группу соединений, изложенное в формуле изобретения, с активным ингредиентом лекарственного средства, указанного в Регистрационном удостоверении.
Аналогичный подход будет осуществлен при проверке композиции лекарственного средства с учетом указанного в Регистрационном удостоверении назначения композиции и ее состава, определяющего лекарственную форму.
Что касается понятия первого разрешения и определения дня получения первого разрешения, то исходя из статьи 28 Федерального закона от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств" первым разрешением следует считать Регистрационное удостоверение на впервые регистрируемый в Российской Федерации препарат, выдаваемый сроком на 5 лет, а днем получения первого разрешения следует считать дату регистрации лекарственного средства, указанную в регистрационном удостоверении, т.е. возникновение права на применение, а не получение документа, подтверждающего это право.
Регистрационное удостоверение на впервые регистрируемый в Российской Федерации препарат выдается сроком на 5 лет. Прежде чем получить такое удостоверение препарат, впервые регистрируемый на территории Российской Федерации, должен пройти длительную процедуру клинических исследований. Именно этот срок клинических исследований не позволяет быстро вводить новый препарат в гражданский оборот и требует компенсации в виде продления патента.
Через пять лет оборота нового препарата на рынке необходимо пройти процедуру повторной регистрации и получить бессрочное Регистрационное удостоверение. Это сделано для того, чтобы если в процессе применения препарата в течение 5 лет будут выявлены неучтенные противопоказания, то либо повторная регистрация не состоится, либо это будет учтено в противопоказаниях.
Таким образом, исходя из самой идеи продления патента, на время затраченное для проведение клинических испытаний, только первое регистрационное удостоверение, выданное на 5 лет, может считаться первым разрешением.
Дополнительно необходимо отметить, что в процедуру рассмотрения заявления о продлении срока действия исключительного права на изобретение введена возможность продления срока ответа на запрос дополнительных материалов на срок не превышающий десять месяцев.
2.3. Примеры, позволяющие лучше понять введенные изменения
2.3.1. Рассмотрение возможности выдачи дополнительного патента на соединение, фармацевтическую композицию и фармацевтики приемлемые соли
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Соединение, которое представляет собой 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол или его фармацевтически приемлемые соли.
2. Фармацевтическая композиция, обладающая активностью антагониста орексинового рецептора, содержащая инертный носитель и соединение по п. 1 или его фармацевтически приемлемую соль.
3. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в медицине.
4. Применение соединения по п. 1 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики нарушения сна.
5. Соединение по п. 1, где соединение представляет собой 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол.
6. Фармацевтическая композиция, обладающая активностью антагониста орексинового рецептора, содержащая фармацевтически приемлемый носитель и соединение 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол или его фармацевтически приемлемую соль.
7. Фармацевтическая композиция по п. 6, где композиция содержит 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых и зависимых пп. 1 - 2 формулы изобретения указанного патента и выдачи дополнительного патента.
Действующим законодательством на момент его подачи является: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, включающим доводы заявителя, на 3 л;
2) Скорректированная формула изобретения для дополнительного патента (с комментариями и без) на 2 л;
3) Копия Регистрационного удостоверения (далее - РУ) N ЛП-000001 от 01.01.2001 на 2 л;
4) Копия Инструкции по применению лекарственного препарата для медицинского применения Б*** (далее - Инструкция) на 16 л;
5) Приложение - информация о структуре лекарственного средства, Реестр ВОЗ, (далее - Приложение) на 2 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков:
1. Соединение, которое представляет собой 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол или его фармацевтически приемлемые соли.
2. Фармацевтическая композиция, обладающая активностью антагониста орексинового рецептора, содержащая инертный носитель и соединение по п. 1 или его фармацевтически приемлемую соль.
Представленная формула дополнительного патента составлена на основании запатентованной формулы изобретения RU***.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству Б***, патентообладатель представляет Регистрационное удостоверение N ЛП-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата Б***, указанная в представленном РУ N ЛП-004058 от 29.12.2016 включает следующие признаки:
Торговое наименование: Б***.
Международное непатентованное, или группировочное, или химическое наименование: Суворексант.
Качественный состав и количественный состав действующих веществ и качественный состав вспомогательных веществ: суворексант 10.00/15.00/20.00 мг, вспомогательные вещества (коповидон, лактозы моногидрат, целлюлоза микрокристаллическая, кроскармеллоза натрия, магния стеарат, пленочная оболочка - Опадрай II зеленый 39К110000 [лактозы моногидрат, гипромеллоза 2910, титана диоксид, триацетин, краситель бриллиантовый голубой алюминиевый лак, железа оксид желтый]/Опадрай II белый 39К18561 [лактозы моногидрат, гипромеллоза 2910, титана диоксид, триацетин]/Опадрай II белый 39К18561 [лактозы моногидрат, гипромеллоза 2910, титана диоксид, триацетин]).
Форма выпуска: таблетки, покрытые пленочной оболочкой, 10 мг, 15 мг, 20 мг (блистер) 10 x 1/3 (пачка картонная).
Назначение: снотворное средство. Лечение бессонницы, характеризующейся затруднениями при засыпании и/или поддержании сна (Инструкция, стр. 2 раздел "Фармакотерапевтическая группа", стр. 5 раздел "Показания к применению").
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если:
- в формуле изобретения продукт охарактеризован в виде соединения или группы соединений, описываемых общей структурной формулой, и из описания изобретения следует возможность его использования в качестве активного ингредиента лекарственного средства, пестицида или агрохимиката, при этом совокупность признаков, определяющих объем правовой охраны продукта, характеризующих соединение или группу соединений, описываемых общей структурной формулой, идентична активному ингредиенту лекарственного средства, пестицида или агрохимиката, указанного в полученном разрешении на применение этого продукта, а описание изобретения содержит информацию о том, что соединение или группа соединений, описываемых общей структурной формулой, обладает такой активностью, которая позволяет его использовать в указанном в разрешении лекарственном средстве, пестициде или агрохимикате;
- в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
1) Сравнение характеристики соединения по независимому п. 1 формулы дополнительного патента и характеристики активного ингредиента лекарственного средства Б*** показывает следующее.
В независимом п. 1 формулы изобретения дополнительного патента охарактеризовано соединение 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол в свободной форме или в форме его фармацевтически приемлемых солей. При этом в описании патента RU*** указано, что данное соединение является антагонистом орексиновых рецепторов и может применяться для лечения или профилактики нарушений сна.
В представленных патентообладателем РУ (см. раздел "МНН"), Инструкции (см. разделы "Состав", "Показания к применению") и Приложении (информация о структуре суворексанта) охарактеризована фармацевтическая композиция для лечения бессонницы, включающая в качестве активного агента соединение суворексант, структурная формула которого совпадает со структурной формулой соединения 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол. Однако возможность использования указанного соединения в форме фармацевтически приемлемых солей в представленных документах не охарактеризована.
Таким образом, независимый п. 1 представленной патентообладателем формулы дополнительного патента не характеризует продукт, относящийся к лекарственному средству Б***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих соединение, не идентична активному ингредиенту лекарственного средства, указанного в полученном разрешении на применение этого продукта, за исключением варианта, когда соединение по п. 1 представляет собой непосредственно 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол.
2) Сравнение характеристики композиции по независимому п. 2 формулы дополнительного патента и характеристики композиции лекарственного средства Б*** показывает следующее.
В п. 2 охарактеризована фармацевтическая композиция, обладающая активностью антагониста орексинового рецептора, которая включает соединение 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол или его фармацевтически приемлемую соль и инертный носитель.
В РУ (см. разделы "МНН", "Качественный состав и количественный состав действующих веществ и качественный состав вспомогательных веществ"), Инструкции (см. раздел "Состав") и Приложении охарактеризована фармацевтическая композиция включающая в качестве действующего агента суворексант (соединение 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол) и инертные наполнители. При этом суворексант является антагонистом орексиновых рецепторов (Инструкция, раздел "Фармакологические свойства").
Между тем, фармацевтически приемлемые соли 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-и}-1,3-бензоксазола в представленных документах не охарактеризованы.
Таким образом, независимый п. 2 представленной патентообладателем формулы дополнительного патента не характеризует продукт, относящийся к лекарственному средству Б***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, не идентична характеристике композиции лекарственного средства, указанного в разрешении, за исключением варианта, когда в качестве активного агента используется 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол.
На основании изложенного дополнительный патент с формулой, представленной патентообладателем совместно с ходатайством (заявление) о продлении срока действия исключительного права на изобретение не может быть выдан.
Дополнительный патент может быть выдан на следующую совокупность признаков:
1. Соединение, которое представляет собой 5-хлор-2-{(5R)-5-метил-4-[5-метил-2-(2H-1,2,3-триазол-2-ил)бензоил]-1,4-диазепан-1-ил}-1,3-бензоксазол.
2. Фармацевтическая композиция, обладающая активностью антагониста орексинового рецептора, содержащая инертный носитель и соединение по п. 1.
2.3.2. Рассмотрение возможности выдачи дополнительного патента на гипертонический раствор и диапазон веществ
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Гипертонический раствор для очищения толстой кишки у млекопитающего, включающий от 80 г до 150 г на литр полиэтиленгликоля (ПЭГ), от 2 г до 15 г на литр сульфата щелочного или щелочноземельного металла или смеси сульфатов щелочного или щелочноземельного металла, электролиты, выбранные из группы электролитов, включающей хлорид натрия и хлорид калия, и от 3 г до 20 г на литр аскорбиновой кислоты и/или, по меньшей мере, одной ее соли или смеси аскорбиновой кислоты и, по меньшей мере одной соли аскорбиновой кислоты.
2. Раствор по п. 1, в котором, по меньшей мере, один сульфат щелочного или щелочноземельного металла представлен в количестве 7,5 г на литр.
3. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 2000 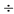 4500 дальтон.
4500 дальтон.
4. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 3350 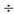 4000 дальтон и представлен в количестве от 90 г до 125 г на литр.
4000 дальтон и представлен в количестве от 90 г до 125 г на литр.
5. Раствор по п. 1, в котором в смеси аскорбиновая кислота и, по меньшей мере, одна соль аскорбиновой кислоты представлены в соотношении от 2:8 до 8:2 и в количестве от 5 г до 15 г на литр.
6. Раствор по п. 1, в котором соль аскорбиновой кислоты выбрана из группы солей, включающей аскорбат натрия, аскорбат калия, аскорбат магния и аскорбат кальция.
7. Раствор по п. 1, в котором хлорид натрия представлен в количестве от 1 г до 5 г на литр.
8. Раствор по п. 1, в котором хлорид калия представлен в количестве от 0,2 г до 4 г на литр.
9. Раствор по п. 1, который дополнительно содержит, по меньшей мере, одну вкусовую добавку.
10. Раствор по п. 1, который дополнительно содержит, по меньшей мере, один подсластитель.
11. Раствор по п. 1, который содержит лимонную кислоту.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых и зависимых пп. 1 - 11 формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, включающим доводы заявителя на 10 л;
2) Копия Регистрационного удостоверения (РУ) N ЛП-000001 от 01.01.2001 на 2 л;
3) Копия Инструкции по применению лекарственного препарата для медицинского применения М*** (далее - Инструкция) на 8 л;
4) Приложение 1 - копия статьи с сайта http://gastroscan.ru о синонимичности понятий "макрогол" и "полиэтиленгликоль" на 3 л.;
5) Приложение 2 - копия публикации Британской комиссии по вопросам Фармакопеи о международном непатентованном названии "макрагол" на 3 л.;
6) Приложение 3 - копия статьи с сайта http://novostioede.ru о синонимичности понятий "ацесульфам калия" и "ацесульфам K" на 2 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков:
1. Гипертонический раствор для очищения толстой кишки у млекопитающего, включающий от 80 г до 150 г на литр полиэтиленгликоля (ПЭГ), от 2 г до 15 г на литр сульфата щелочного или щелочноземельного металла или смеси сульфатов щелочного или щелочноземельного металла, электролиты, выбранные из группы электролитов, включающей хлорид натрия и хлорид калия, и от 3 г до 20 г на литр аскорбиновой кислоты и/или, по меньшей мере, одной ее соли или смеси аскорбиновой кислоты и, по меньшей мере одной соли аскорбиновой кислоты.
2. Раствор по п. 1, в котором, по меньшей мере, один сульфат щелочного или щелочноземельного металла представлен в количестве 7,5 г на литр.
3. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 2000 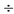 4500 дальтон.
4500 дальтон.
4. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 3350  4000 дальтон и представлен в количестве от 90 г до 125 г на литр.
4000 дальтон и представлен в количестве от 90 г до 125 г на литр.
5. Раствор по п. 1, в котором в смеси аскорбиновая кислота и, по меньшей мере, одна соль аскорбиновой кислоты представлены в соотношении от 2:8 до 8:2 и в количестве от 5 г до 15 г на литр.
6. Раствор по п. 1, в котором соль аскорбиновой кислоты выбрана из группы солей, включающей аскорбат натрия, аскорбат калия, аскорбат магния и аскорбат кальция.
7. Раствор по п. 1, в котором хлорид натрия представлен в количестве от 1 г до 5 г на литр.
8. Раствор по п. 1, в котором хлорид калия представлен в количестве от 0,2 г до 4 г на литр.
9. Раствор по п. 1, который дополнительно содержит, по меньшей мере, одну вкусовую добавку.
10. Раствор по п. 1, который дополнительно содержит, по меньшей мере, один подсластитель.
11. Раствор по п. 1, который содержит лимонную кислоту.
Формула дополнительного патента идентичная формуле запатентованного изобретения RU***.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству М***, патентообладатель представляет Регистрационное удостоверение N ЛП-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата М***, указанная в представленном разрешении ЛП-00001 от 01.01.2001, включает следующие признаки:
Торговое наименование лекарственного препарата: М***.
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: отсутствует.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): саше А: макрогол-3350 100.00 г, натрия сульфат 7.500 г, натрия хлорид 2.691 г, калия хлорид 1.015 г, вспомогательные вещества (аспартам (Е951) 0.233 г, ацесульфам калия 0.117 г, ароматизатор лимонный V3938-1 N 1 0.340 г); саше Б: аскорбиновая кислота 4.700 г, натрия аскорбат 5.900 г.
Форма выпуска: порошок для приготовления раствора для приема внутрь (саше А) 111.896 г x 2 + (саше Б) 10.600 г x 2 (пачка картонная).
Назначение: слабительное средство, подготовка к диагностическим исследованиям и оперативным вмешательствам, требующим опорожнения кишечника (Инструкция, стр. 1 раздел "Фармакотерапевтическая группа", стр. 2 раздел "Показания к применению").
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Сравнение характеристики композиции по независимому п. 1 формулы дополнительного патента и характеристики композиции лекарственного средства М*** показывает следующее.
В независимом п. 1 формулы изобретения дополнительного патента охарактеризован гипертонический раствор для очищения толстой кишки у млекопитающего. Указанный раствор включает следующие компоненты: от 80 до 150 г/л полиэтиленгликоля, от 2 до 15 г/л сульфата щелочного или щелочноземельного металла или смеси сульфатов щелочного или щелочноземельного металла, электролиты, выбранные из хлорида натрия и хлорида калия, а также от 3 до 20 г/л аскорбиновой кислоты и/или, по меньшей мере, одной ее соли, или смеси аскорбиновой кислоты и, по меньшей мере, одной ее соли. Из вышеизложенного следует, что компонентный состав раствора в п. 1 охарактеризован с использованием альтернативных понятий, а количественные признаки выражены в виде интервала значений. Таким образом, совокупность признаков п. 1 предполагает несколько вариантов качественного и количественного состава ингредиентов заявленного раствора.
Между тем, в представленных РУ (см. раздел "Состав лекарственного средства") и Инструкции (см. разделы "Состав", "Фармакологические свойства"), а также представленных патентообладателем приложениях охарактеризован раствор для очищения толстой кишки, включающий 100.00 г/л макрогола-3350 (синоним полиэтиленгликоля), 7.500 г/л натрия сульфата (сульфат щелочного металла), 2.691 г/л натрия хлорида и 1.015 г/л калия хлорида (электролиты), 4.700 г/л аскорбиновой кислоты и 5.900 г/л натрия аскорбата (т.е. 10.600 г/л смеси аскорбиновой кислоты и ее соли), а также 0.233 г/л аспартама (E951), 0.117 г/л ацесульфама калия и 0.340 г/л ароматизатора лимонный V3938-1 N 1. В результате расчета осмолярности раствора Мовипреп с учетом указанных количеств ингредиентов можно сделать вывод, что данный раствор является гипертоническим.
Таким образом, в РУ, Инструкции и приложениях охарактеризован только один конкретный вариант качественного и количественного состава заявленного раствора. Указанные в п. 1 дополнительного патента альтернативы "сульфат щелочноземельного металла", "смесь сульфатов щелочного металла", "смесь сульфатов щелочноземельного металла", "аскорбиновая кислота", "по меньшей мере, одна соль аскорбиновой кислоты" в РУ и Инструкции не охарактеризованы. Кроме того, содержание полиэтиленгликоля, сульфата натрия и смеси аскорбиновой кислоты и ее соли в препарате М*** выражено в виде единичных значений - 100 г/л, 7,5 г/л и 10,6 г/л соответственно. Каждое из данных значений соответствует только одному из возможных точечных вариантов, входящих в количественные интервалы, которые указанны в п. 1 дополнительного патента. В РУ и Инструкции не охарактеризована возможность включения полиэтиленгликоля в раствор в количестве 80 - 150 г/л, за исключением 100 г/л, сульфата щелочного металла - в количестве 2 - 15 г/л, за исключением 7,5 г/л, и смеси аскорбиновой кислоты и ее соли - в количестве 3 - 20 г/л, за исключением 10,6 г/л.
Таким образом, независимый п. 1 представленной патентообладателем формулы дополнительного патента не характеризует продукт, относящийся к лекарственному средству М***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, не идентична характеристике композиции лекарственного средства, указанного в разрешении, за исключением одного варианта состава, указанного выше.
Относительно зависимых п. п. 2 - 11 дополнительного патента необходимо отметить следующее.
Признаки зависимого п. 2, касающиеся содержания сульфата щелочного или щелочноземельного металла в количестве 7,5 г на литр, охарактеризованы в РУ только в отношении сульфата щелочного металла (раздел "Состав лекарственного средства", компонент сульфат натрия). Возможность включения в раствор сульфата щелочноземельного металла в количестве 7,5 г/л в РУ и Инструкции не охарактеризована. Таким образом, заявитель вправе исключить из п. 2 альтернативу "щелочноземельного металла", которая не охарактеризована в РУ.
Признаки зависимых пп. 3 - 4, касающиеся молекулярного веса полиэтиленгликоля, изложенные в виде широких диапазонов значений "2000 - 4500 дальтон" и "3350 - 4000 дальтон" соответственно, не охарактеризованы в РУ и Инструкции во всех возможных вариантах, за исключением единичного значения "3350 дальтон" (см. раздел "Состав лекарственного средства"). В связи с этим заявитель вправе уточнить пп. 3 - 4 в части молекулярного веса полиэтиленгликоля до конкретного значения "3350 дальтон", которое входит в заявленные по пп. 3 - 4 количественные интервалы и охарактеризовано в РУ. Кроме того, заявитель вправе уточнить п. 4 в части количественного содержания полиэтиленгликоля как "представлен в количестве 100 г на литр", поскольку это единственное из значений, входящих в указанный в п. 4 диапазон "от 90 до 125 г на литр", которое охарактеризовано в РУ (раздел "Состав лекарственного средства").
Признаки зависимого п. 5, касающиеся соотношения аскорбиновой кислоты и ее соли в смеси, а также количественного содержания данной смеси в растворе, изложенные в виде широких диапазонов значений "в соотношении от 2:8 до 8:2" и "в количестве от 5 г до 15 г на литр", не охарактеризованы в РУ и Инструкции во всех возможных вариантах, за исключением единичных значений "в соотношении 4,7:5,9" и "в количестве 10,6 г на литр" (см. раздел "Состав лекарственного средства"). В связи с этим заявитель вправе уточнить п. 5 в части соотношения и количественного содержания смеси аскорбиновой кислоты и ее соли до конкретных значений "4,7:5,9" и "10,6 г на литр", которые входят в заявленные по п. 5 интервалы и охарактеризованы в РУ.
Признаки зависимого п. 6, касающиеся вида соли аскорбиновой кислоты, не охарактеризованы в РУ и Инструкции во всех возможных альтернативах, за исключением варианта "аскорбат натрия" (см. РУ раздел "Состав лекарственного средства").
Признаки зависимого п. 7, касающиеся количественного содержания хлорида натрия, изложенные в виде широкого диапазона значений "от 1 г до 5 г на литр", не охарактеризованы в РУ и Инструкции во всех возможных вариантах, за исключением единичного значения "2,691 г на литр" (см. раздел "Состав лекарственного средства"). В связи с этим заявитель вправе уточнить п. 7 в части количественного содержания хлорида натрия до конкретного значения "2,691 г на литр", которое входит в заявленный по п. 7 количественный интервал и охарактеризовано в РУ.
Признаки зависимого п. 8, касающиеся количественного содержания хлорида калия, изложенные в виде широкого диапазона значений "от 0,2 г до 4 г на литр", не охарактеризованы в РУ и Инструкции во всех возможных вариантах, за исключением единичного значения "1,015 г на литр" (см. раздел "Состав лекарственного средства"). В связи с этим заявитель вправе уточнить п. 8 в части количественного содержания хлорида калия до конкретного значения "1,015 г на литр", которое входит в заявленный по п. 8 количественный интервал и охарактеризовано в РУ.
Признаки зависимого п. 9, касающиеся содержания в растворе вкусовой добавки, охарактеризованы в РУ (раздел "Состав лекарственного средства", компонент ароматизатор лимонный V3938-1 N 1).
Признаки зависимого п. 10, касающиеся содержания в растворе подсластителей, охарактеризованы в РУ (раздел "Состав лекарственного средства", компоненты аспартам и ацесульфам калия).
Признаки п. 11, касающиеся содержания в растворе лимонной кислоты, не охарактеризованы в РУ и Инструкции.
С учетом вышеизложенного основания для выдачи дополнительного патента с формулой, представленной заявителем совместно с ходатайством отсутствуют.
Дополнительный патент может быть выдан на следующую совокупность признаков:
1. Гипертонический раствор для очищения толстой кишки у млекопитающего, включающий 100 г на литр полиэтиленгликоля (ПЭГ), 7,5 г на литр сульфата щелочного металла, электролиты, выбранные из группы электролитов, включающей хлорид натрия и хлорид калия, и 10,6 г на литр смеси аскорбиновой кислоты и ее соли.
2. Раствор по п. 1, в котором сульфат щелочного металла представлен в количестве 7,5 г на литр.
3. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 3350 дальтон.
4. Раствор по п. 1, в котором ПЭГ имеет молекулярный вес 3350 дальтон и представлен в количестве 100 г на литр.
5. Раствор по п. 1, в котором в смеси аскорбиновая кислота и соль аскорбиновой кислоты представлены в соотношении 4,7:5,9 и в количестве 10,6 г на литр.
6. Раствор по п. 1, в котором соль аскорбиновой кислоты выбрана из аскорбата натрия.
7. Раствор по п. 1, в котором хлорид натрия представлен в количестве 2,691 г на литр.
8. Раствор по п. 1, в котором хлорид калия представлен в количестве 1,015 г на литр.
9. Раствор по п. 1, который дополнительно содержит, по меньшей мере, одну вкусовую добавку.
10. Раствор по п. 1, который дополнительно содержит, по меньшей мере, один подсластитель.
2.3.3. Рассмотрение возможности выдачи патента на фармацевтическую композицию, общее - частное
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Композиция для топического нанесения, предназначенная для использования человеком, отличающаяся тем, что она представляет собой эмульсию, содержащую:
a) масляную фазу, содержащую жиры, выбранные из изопропилпальмитата, цетилового спирта, стеарилового спирта, диметикона 200 20 cs;
b) поверхностно-активный эмульгатор, выбранный из Цетеарета-20, сорбитанмоностеарата;
c) ивермектин;
d) смесь растворителей и/или пропенетрантов действующего начала, выбранных из пропиленгликоля, олеилового спирта, феноксиэтанола;
e) и воду.
2. Композиция по п. 1, отличающаяся тем, что она содержит:
a) масляную фазу, содержащую жиры, выбранные из изопропилпальмитата, цетилового спирта, стеарилового спирта, диметикона 200 20 cs;
b) поверхностно-активный эмульгатор, выбранный из Цетеарета-20, сорбитанмоностеарата;
c) ивермектин;
d) смесь растворителей и/или пропенетрантов действующего начала, выбранных из пропиленгликоля, олеилового спирта, феноксиэтанола;
e) желирующий агент Акрилат/С10-30-алкилакрилатный кроссполимер;
f) и воду.
3. Композиция по одному из п. 1 или 2, отличающаяся тем, что она содержит в воде:
|
Ивермектин
|
1,00
|
|
Глицерин
|
4,0
|
|
Акрилат/С10-30-алкилакрилатный кроссполимер
|
0,2
|
|
Метил-п-гидроксибензоат
|
0,2
|
|
Двунатриевая ЭДТА
|
0,05
|
|
Моногидрат лимонной кислоты
|
0,05
|
|
Изопропилпальмитат
|
4,0
|
|
Цетиловый спирт
|
3,5
|
|
Стеариловый спирт
|
2,5
|
|
Олеиловый спирт
|
2,0
|
|
Цетеарет-20
|
3,0
|
|
Сорбитанмоностеарат
|
2,0
|
|
Диметикон 200 20 cs
|
0,5
|
|
Пропил-п-гидроксибензоат
|
0,1
|
|
Пропиленгликоль
|
2,0
|
|
Феноксиэтанол
|
1,0
|
|
Гидроксид натрия 10%
|
qs
|
|
для pH
|
|
|
Вода
|
qs до
|
|
100
|
в мас.% по отношению к общей массе композиции, и представляет собой крем.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых и зависимых пп. 1 - 3 формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, Дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, включающим доводы заявителя на 6 л;
2) Скорректированная формула изобретения для дополнительного патента (с комментариями и без) на 6 л;
3) Копия Регистрационного удостоверения (РУ) N ЛП-000001 от 01.01.2001 на 2 л;
4) Копия Инструкции по применению лекарственного препарата для медицинского применения С*** (далее - Инструкция) на 3 л;
5) Приложение 1 - Справочная литература, из которой известно, что акрилат/С10-С30 алкил акрилатный кроссполимер является карбомером, на 1 л.;
6) Приложение 2 (характеристика динатрия эдетата) - Государственная Фармакопея СССР, "Медицина", Москва, 1986, стр. 798;
7) Приложение 3 - Справочная литература, из которой известен состав Цетеарета-20, а также раскрыта характеристика полиэтиленгликоля, на 2 л.;
8) Приложение 4 - Справочная литература, где раскрывается, что заболевание розацеа представляет собой розовые угри, на 1 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков:
1. Композиция для топического нанесения, предназначенная для использования человеком, отличающаяся тем, что она представляет собой эмульсию, содержащую:
a) масляную фазу, содержащую жиры, выбранные из изопропилпальмитата, цетилового спирта, стеарилового спирта, диметикона 200 20 cs;
b) поверхностно-активный эмульгатор, выбранный из Цетеарета-20, сорбитанмоностеарата;
c) ивермектин;
d) смесь растворителей и/или пропенетрантов действующего начала, выбранных из пропиленгликоля, олеилового спирта, феноксиэтанола;
e) и воду.
2. Композиция по п. 1, отличающаяся тем, что она содержит:
a) масляную фазу, содержащую жиры, выбранные из изопропилпальмитата, цетилового спирта, стеарилового спирта, диметикона 200 20 cs;
b) поверхностно-активный эмульгатор, выбранный из Цетеарета-20, сорбитанмоностеарата;
c) ивермектин;
d) смесь растворителей и/или пропенетрантов действующего начала, выбранных из пропиленгликоля, олеилового спирта, феноксиэтанола;
e) желирующий агент Акрилат/С10-30-алкилакрилатный кроссполимер;
f) и воду.
3. Композиция по одному из п. 1 или 2, отличающаяся тем, что она представляет собой крем и содержит в воде:
|
Ивермектин
|
1,00
|
|
Глицерин
|
4,0
|
|
Акрилат/С10-30-алкилакрилатный кроссполимер
|
0,2
|
|
Метил-п-гидроксибензоат
|
0,2
|
|
Двунатриевая ЭДТА
|
0,05
|
|
Моногидрат лимонной кислоты
|
0,05
|
|
Изопропилпальмитат
|
4,0
|
|
Цетиловый спирт
|
3,5
|
|
Стеариловый спирт
|
2,5
|
|
Олеиловый спирт
|
2,0
|
|
Цетеарет-20
|
3,0
|
|
Сорбитанмоностеарат
|
2,0
|
|
Диметикон 200 20 cs
|
0,5
|
|
Пропил-п-гидроксибензоат
|
0,1
|
|
Пропиленгликоль
|
2,0
|
|
Феноксиэтанол
|
1,0
|
|
Гидроксид натрия 10%
|
qs
|
|
для pH
|
|
|
Вода
|
qs до
|
|
100
|
в мас.% по отношению к общей массе композиции.
Представленная формула дополнительного патента составлена патентообладателем на основании запатентованной формулы изобретения RU***.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству С***, патентообладатель представляет Регистрационное удостоверение N ЛП-00001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата С***, указанная в представленном разрешении ЛП-000001 от 01.01.2001 включает следующие признаки:
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: Ивермектин.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): ивермектин 10.0 мг, вспомогательные вещества (глицерол 40.0 мг, изопропилпальмитат 40.0 мг, карбомер сополимер тип В 2.0 мг, диметикон 20 Cst 5.0 мг, динатрия эдетат 0.5 мг, лимонной кислоты моногидрат 0.5 мг, цетиловый спирт 35.0 мг, стеариловый спирт 25.0 мг, макрогола цетостеариловый эфир 30.0 мг, сорбитана стеарат 20.0 мг, метилпарагидроксибензоат 2.0 мг, пропилпарагидроксибензоат 1.0 мг, феноксиэтанол 10.0 мг, пропиленгликоль 20.0 мг, олеиловый спирт 20.0 мг, натрия гидроксида раствор 10% до pH 6,3 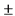 0.3, вода очищенная до 1000 мг).
0.3, вода очищенная до 1000 мг).
Форма выпуска: крем для наружного применения, 1% (туба) 15/30 г x 1 (пачка картонная).
Назначение: противомикробное и противопротозойное средство. Лечение воспалительных поражений кожи при розацеа (папуло-пустулезная форма) у взрослых пациентов (Инструкция, стр. 1 раздел "Фармакотерапевтическая группа", стр. 3 раздел "Показания").
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Сравнение характеристики композиции по независимому п. 1 формулы дополнительного патента и характеристики композиции лекарственного средства С*** показывает следующее.
В независимом п. 1 охарактеризована композиция для топического нанесения, предназначенная для использования человеком. Указанная композиция содержит ивермектин в качестве активного агента и следующие вспомогательные вещества: жиры, выбранные из изопропилпальмитата, цетилового спирта, стеарилового спирта, диметикона 200 20 cs; поверхностно-активный эмульгатор, выбранный из Цетеарета-20, сорбитанмоностеарата; смесь растворителей и/или пропенетрантов действующего начала, выбранных из пропиленгликоля, олеилового спирта, феноксиэтанола; воду. То есть совокупность признаков, характеризующих содержание ингредиентов композиции по п. 1, предполагает возможность большого количества вариантов ее качественного и количественного состава.
В представленных РУ (см. разделы "МНН", "Состав лекарственного средства") и Инструкции (см. разделы "Состав", "Показания"), а также представленных патентообладателем приложениях 1, 2, 3 и 4, охарактеризована фармацевтическая композиция для лечения розацеи, включающая конкретные компоненты в строго определенных количествах, а именно: ивермектин в количестве 10.0 мг, а также вспомогательные вещества: глицерол 40.0 мг, изопропилпальмитат 40.0 мг, карбомер сополимер тип В 2.0 мг, диметикон 20 Cst 5.0 мг, динатрия эдетат 0.5 мг, лимонной кислоты моногидрат 0.5 мг, цетиловый спирт 35.0 мг, стеариловый спирт 25.0 мг, макрогола цетостеариловый эфир 30.0 мг, сорбитана стеарат 20.0 мг, метилпарагидроксибензоат 2.0 мг, пропилпарагидроксибензоат 1.0 мг, феноксиэтанол 10.0 мг, пропиленгликоль 20.0 мг, олеиловый спирт 20.0 мг, натрия гидроксида раствор 10% до pH 6,3 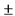 0.3, вода очищенная до 1000 мг. То есть в РУ и указанных выше дополнительных материалах охарактеризован только один из возможных вариантов композиции по п. 1, который раскрыт в зависимом п. 3 дополнительного патента. Все остальные альтернативные составы композиции по п. 1 не охарактеризованы в РУ, Инструкции и приложениях.
0.3, вода очищенная до 1000 мг. То есть в РУ и указанных выше дополнительных материалах охарактеризован только один из возможных вариантов композиции по п. 1, который раскрыт в зависимом п. 3 дополнительного патента. Все остальные альтернативные составы композиции по п. 1 не охарактеризованы в РУ, Инструкции и приложениях.
Кроме того, в п. 1 дополнительного патента указано, что фармацевтическая композиция представлена в форме эмульсии, тогда как согласно РУ (см. раздел "Форма выпуска") зарегистрированное лекарственное средство Солантра представляет собой крем. Эмульсия - это однородная по внешнему виду лекарственная форма, состоящая из взаимно нерастворимых тонкодиспергированных жидкостей, предназначенная для внутривенного, наружного или парентерального применения. Крем - это мягкая лекарственная форма для местного применения, представляющая собой двух- или многофазную дисперсную систему, где дисперсионная среда при установленной температуре хранения имеет ньютоновский тип течения и низкие значения реологических параметров, крем получают на основе эмульсии, стабилизированной подходящими эмульгаторами. Из вышеизложенных определений следует, что "крем" и "эмульсия" не идентичные понятия. Крем является частным случаем эмульсионной лекарственной формы.
Таким образом, независимый п. 1 представленной патентообладателем формулы дополнительного патента не характеризует продукт, относящийся к лекарственному средству С***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, не идентична характеристике композиции лекарственного средства, указанного в разрешении, за исключением одного варианта конкретного состава, охарактеризованного в зависимом п. 3 формулы изобретения дополнительного патента, и лекарственной формы "крем".
Относительно зависимого п. 2 следует отметить следующее. Совокупность признаков зависимого п. 8 предполагает несколько альтернативных составов композиции, тогда как лекарственное средство С*** соответствует лишь одному из них (см. РУ, раздел "Состав лекарственного средства"). Таким образом, совокупность признаков зависимого п. 2 не охарактеризована в представленных патентообладателем регистрационных документах во всех возможных вариантах, за исключением одного конкретного качественного и количественного состава ингредиентов композиции, охарактеризованного в зависимом п. 3 дополнительного патента.
Совокупность признаков зависимого п. 3, касающихся конкретного состава ингредиентов композиции, охарактеризована в РУ (см. раздел "Состав лекарственного средства") и Инструкции (раздел "Состав").
С учетом вышеизложенного основания для выдачи дополнительного патента с формулой, представленной заявителем совместно с ходатайством отсутствуют.
Дополнительный патент может быть выдан на следующую совокупность признаков:
1. Композиция для топического нанесения в виде крема, предназначенная для использования человеком, предназначенной для лечения розовых угрей, причем композиция содержит в воде:
|
Ивермектин
|
1,00
|
|
Глицерин
|
4,0
|
|
Акрилат/Сю-зо-алкилакрилатный кроссполимер
|
0,2
|
|
Метил-п-гидроксибензоат
|
0,2
|
|
Двунатриевая ЭДТА
|
0,05
|
|
Моногидрат лимонной кислоты
|
0,05
|
|
Изопропилпальмитат
|
4,0
|
|
Цетиловый спирт
|
3,5
|
|
Стеариловый спирт
|
2,5
|
|
Олеиловый спирт
|
2,0
|
|
Цетеарет-20
|
3,0
|
|
Сорбитанмоностеарат
|
2,0
|
|
Диметикон 200 20 cs
|
0,5
|
|
Пропил-п-гидроксибензоат
|
0,1
|
|
Пропиленгликоль
|
2,0
|
|
Феноксиэтанол
|
1,0
|
|
Гидроксид натрия 10%
|
qs
|
|
для pH
|
|
|
Вода
|
qs до
|
|
100
|
в мас.% по отношению к общей массе композиции.
2.3.4. Рассмотрение возможности выдачи дополнительного патента на фармацевтическую композицию, в которой формула дополнительного патента содержит признаки, отсутствующие в описании и формуле первоначального патента
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков
1. Композиция, содержащая аклидиний для лечения или профилактики респираторного заболевания или состояния у пациента посредством ингаляции, при этом у указанных пациентов не наблюдаются системные антимускариновые действия, где пациент страдает от состояния или чувствителен к нему, которое может обостряться при действии системной антимускариновой активности.
2. Композиция по п. 1, где респираторное заболевание или состояние выбраны из острого или хронического бронхита, эмфиземы, астмы и хронического обструктивного заболевания легких, прежде всего астмы и хронического обструктивного заболевания легких.
3. Композиция по любому из предшествующих пунктов, где пациент страдает от состояния или чувствителен к одному или более состояний, выбранных из следующих состояний:
1) шизофрения, нарушение концентрации внимания, спутанность сознания, возбужденное состояние, делирий, нарушение внимания, нарушение памяти, угнетение дыхания,
2) глаукома, сухость глаз, увеличенный размер зрачка, ухудшение остроты зрения, повышенное глазное давление,
3) увеличенная предстательная железа или ее непроходимость, затрудненное мочеиспускание,
4) сужение или непроходимость тонкой кишки, увеличение ободочной кишки, хронический запор, расширение нижнего отдела пищевода, сниженная перистальтика желудка, запор,
5) сухость во рту, раздражение в горле, нарушенное потоотделение,
6) сердечно-сосудистые заболевания (включая любой рестеноз, артериосклероз, предшествующие инсульт или сердечный приступ, застойную сердечную недостаточность), аритмия, тахикардия,
7) болезнь Паркинсона, болезнь Альцгеймера, деменция и/или
8) тяжелая псевдопаралитическая миастения.
4. Композиция по любому из предшествующих пунктов, где:
- пациентом является мужчина; и/или
- возраст пациента составляет более шестидесяти лет; и/или
- пациент водит машину или работает с оборудованием в течение курса лечения; и/или
- пациенту вводят второе лекарственное средство, которое является системным активным антихолинергическим агентом, который может вызвать любое состояние по п. 3 или привести к обострению указанных состояний, где второе лекарственное средство выбрано из атипических нейролептиков, трициклических антидепрессантов и антигистаминов.
5. Композиция по п. 4, где пациенту вводят лекарственное средство, которое может усиливать функцию ацетилхолина.
6. Композиция по любому из предшествующих пунктов, где аклидиний представлен в форме бромида аклидиния.
7. Композиция по любому из предшествующих пунктов, где аклидиний представлен в форме сухого порошка, пригодного для ингаляции.
8. Композиция по любому из предшествующих пунктов, где пациенту вводят одно или более дополнительных лекарственных средств для лечения респираторного заболевания или состояния.
9. Композиция по п. 8, где дополнительное лекарственное средство для лечения респираторного заболевания или состояния выбрано из бета-адренергических агонистов, кортикостероидов или глюкокортикостероидов, ингибиторов ФДЕ IV, антигистаминов, антител против IgE, ингибиторов лейкотриена D4, ингибиторов киназы EGFR, ингибиторов киназы p38 и/или антагонистов рецептора NK1.
10. Композиция по п. 9, где дополнительное лекарственное средство выбрано из кортикостероидов и бета-адренергических агонистов.
11. Фармацевтическая композиция для лечения или профилактики посредством ингаляции респираторного заболевания или состояния по любому из пп. 1 или 2 у пациента по любому из пп. 1, 3 - 5 и 8 - 10, и указанная фармацевтическая композиция содержит фармацевтически приемлемый носитель и, в качестве активного агента, аклидиний по любому из пп. 1, 6 и 7.
12. Фармацевтическая композиция по п. 11, где фармацевтически приемлемым носителем является носитель, выбранный из моно-, ди-, и полисахаридов и альдитов, предпочтительно лактоза.
13. Способ лечения или профилактики посредством ингаляции респираторного заболевания или состояния по любому из пп. 1 или 2 у пациента по любому из пп. 1, 3 - 5 и 8 - 10, и указанный способ заключается во введении аклидиния по любому из пп. 1, 6 и 7.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых и зависимых пп. 1 - 4, 6, 7, 11, 12 формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, включающим доводы заявителя, на 3 л;
2) Скорректированная формула изобретения для дополнительного патента (с комментариями и без) на 4 л;
3) Копия Регистрационного удостоверения (РУ) N ЛП-00001 от 01.01.2001 на 2 л;
4) Копия Инструкции по применению лекарственного препарата для медицинского применения Д*** (далее - Инструкция) на 21 л;
5) Копия титульного листа и формулы изобретения патента RU*** на 3 л.
Согласно п. 2 ст. 1363 Кодекса продление срока действия исключительного права на изобретение осуществляется в отношении изобретения, относящегося к лекарственному средству, пестициду или агрохимикату.
Согласно п. 2 ст 1363 Кодекса при продлении срока действия исключительного права выдается дополнительный патент с формулой, содержащей совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков:
1. Композиция содержащая аклидиний для лечения или профилактики респираторного заболевания или состояния у пациента посредством ингаляции, при этом у указанных пациентов не наблюдаются системные антимускариновые действия, где пациент страдает от состояния или чувствителен к нему, которое может обостряться при действии системной антимускариновой активности и где пациенту вводят одно дополнительное лекарственное средство формотерол в свободной форме или в форме фармацевтически приемлемой соли для лечения респираторного заболевания или состояния.
1. Композиция содержащая аклидиний для лечения или профилактики респираторного заболевания или состояния у пациента посредством ингаляции, при этом у указанных пациентов не наблюдаются системные антимускариновые действия, где пациент страдает от состояния или чувствителен к нему, которое может обостряться при действии системной антимускариновой активности и где пациенту вводят одно дополнительное лекарственное средство формотерол в свободной форме или в форме фармацевтически приемлемой соли для лечения респираторного заболевания или состояния.
2. Композиция по п. 1, где респираторное заболевание или состояние выбраны из острого или хронического бронхита, эмфиземы, астмы и хронического обструктивного заболевания легких, прежде всего астмы и хронического обструктивного заболевания легких.
3. Композиция по любому из предшествующих пунктов, где пациент страдает от состояния или чувствителен к одному или более состояний, выбранных из следующих состояний:
1) шизофрения, нарушение концентрации внимания, спутанность сознания, возбужденное состояние, делирий, нарушение внимания, нарушение памяти, угнетение дыхания,
2) глаукома, сухость глаз, увеличенный размер зрачка, ухудшение остроты зрения, повышенное глазное давление,
3) увеличенная предстательная железа или ее непроходимость, затрудненное мочеиспускание,
4) сужение или непроходимость тонкой кишки, увеличение ободочной кишки, хронический запор, расширение нижнего отдела пищевода, сниженная перистальтика желудка, запор,
5) сухость во рту, раздражение в горле, нарушенное потоотделение,
6) сердечно-сосудистые заболевания (включая любой рестеноз, артериосклероз, предшествующие инсульт или сердечный приступ, застойную сердечную недостаточность), аритмия, тахикардия,
7) болезнь Паркинсона, болезнь Альцгеймера, деменция и/или
8) тяжелая псевдопаралитическая миастения.
4. Композиция по любому из предшествующих пунктов, гдепациентом является мужчина; и/или возраст пациента составляет более шестидесяти лет; и/или пациент водит машину или работает с оборудованием в течение курса лечения; и/или пациенту вводят второе лекарственное средство, которое является системным активным антихолинергическим агентом, который может вызвать любое состояние по п. 3 или привести к обострению указанных состояний, где второе лекарственное средство выбрано из атипических нейролептиков, трициклических антидепрессантов и антигистаминов.
5. Композиция по любому из предшествующих пунктов, где аклидиний представлен в форме бромида аклидиния.
6. Композиция по любому из предшествующих пунктов, где аклидиний представлен в форме сухого порошка, пригодного для ингаляции.
7. Фармацевтическая композиция для лечения или профилактики посредством ингаляции респираторного заболевания или состояния по любому из пп. 1 или 2 у пациента по любому из пп. 1 - 6, и указанная фармацевтическая композиция содержит фармацевтически приемлемый носитель и, в качестве активного агента, аклидиний по любому из пп. 1, 5 и 6.
8. Фармацевтическая композиция по п. 7, где фармацевтически приемлемым носителем является носитель, выбранный из моно-, ди-, и полисахаридов и альдитов, предпочтительно лактоза.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы патента RU***, к лекарственному средству Д***, патентообладатель представляет Регистрационное удостоверение N ЛП-0000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата Д***, указанная в представленном разрешении ЛП-000001 от 01.01.2001 включает следующие признаки:
Торговое наименование: Д***.
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: Аклидиния бромид + Формотерол.
Качественный состав и количественный состав действующих веществ и качественный состав вспомогательных веществ: аклидиния бромид микронизированный 0.400 мг [в расчете на аклидиний 0.343 мг], формотерола фумарата дигидрат микронизированный 0.012 мг, вспомогательные вещества (лактозы моногидрат).
Форма выпуска: порошок для ингаляций дозированный, 340 мкг + 11.8 мкг/доза (ингалятор) 60 доз x 1 (пачка картонная).
Назначение: бронходилатирующее средство комбинированное (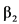 -адреномиметик селективный + м-холиноблокатор). Облегчение симптомов хронической обструктивной болезни легких у взрослых (Инструкция, стр. 1 раздел "Фармакотерапевтическая группа", стр. 7 раздел "Показания к применению").
-адреномиметик селективный + м-холиноблокатор). Облегчение симптомов хронической обструктивной болезни легких у взрослых (Инструкция, стр. 1 раздел "Фармакотерапевтическая группа", стр. 7 раздел "Показания к применению").
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
1) Сравнение характеристики композиции по независимому п. 1 формулы дополнительного патента в части признаков, которые раскрыты в запатентованной формуле RU***, и характеристики композиции лекарственного средства Д*** показывает следующее.
В независимом п. 1 формулы изобретения дополнительного патента охарактеризована композиция, содержащая аклидиний для лечения или профилактики респираторного заболевания или состояния у пациента посредством ингаляции, при этом у указанных пациентов не наблюдаются системные антимускариновые действия, где пациент страдает от состояния или чувствителен к нему, которое может обостряться при действии системной антимускариновой активности и где пациенту вводят одно дополнительное лекарственное средство формотерол в свободной форме или в форме фармацевтически приемлемой соли для лечения респираторного заболевания или состояния.
В представленных патентообладателем РУ (см. разделы "МНН", "Качественный состав и количественный состав действующих веществ и качественный состав вспомогательных веществ") и Инструкции (см. разделы "Состав", "Фармакологические свойства", "Показания к применению") охарактеризовано комбинированное лекарственное средство для облегчения симптомов хронической обструктивной болезни легких (частный случай респираторного заболевания) посредством ингаляции, содержащее в качестве активных веществ аклидиния бромид и формотерола фумарат дигидрат. Указанное средство характеризуется низким количеством системных антихолинергических (антимускариновых) побочных эффектов (см. Инструкция стр. 2 раздел "Фармакологические свойства. Механизм действия") и может с осторожностью применяться у пациентов, страдающих стенокардией, аритмией, перенесших инфаркт миокарда и т.д. (состояния, которые могут обостряться при действии системной антимускариновой активности) (см. Инструкция стр. 7 раздел "С осторожностью").
Из вышеизложенного следует, что охарактеризованное в РУ и Инструкции лекарственное средство Д*** включает два активных компонента в составе одной композиции.
Таким образом, независимый п. 1 представленной патентообладателем формулы дополнительного патента в части признаков, которые раскрыты в запатентованной формуле RU***, характеризует продукт, относящийся к лекарственному средству Д***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, указанного в разрешении.
Несмотря на то, что характеристики фармацевтической композиции представленной в формуле дополнительного патента по п. 1 и характеристики фармацевтической композиции зарегистрированного лекарственного средства идентичны продлить исключительное право по данному патенту не представляется возможным по причинам указанным ниже.
Необходимо обратить внимание на следующее.
В заявлении патентообладатель ходатайствует о продления срока действия патента RU*** на изобретение, охарактеризованное в пп. 1 - 4, 6, 7, 11, 12 запатентованной формулы изобретения RU***. Ни в одном из указанных пунктов не охарактеризовано введение пациенту дополнительного лекарственного средства, которое представляет собой формотерол в свободной форме или в форме фармацевтически приемлемой соли. Таким образом, не может быть признано, что п. 1 формулы изобретения дополнительного патента в части признаков "где пациенту вводят одно дополнительное лекарственное средство формотерол в свободной форме или в форме фармацевтически приемлемой соли для лечения респираторного заболевания или состояния" составлен на основании пунктов запатентованной формулы RU***.
Таким образом, нельзя признать что формула дополнительного патента, представленная патентообладателем совместно с ходатайством составлена на основании запатентованной формулы RU*** (указанной выше).
С учетом вышеизложенного основания для выдачи дополнительного патента с формулой, представленной заявителем совместно с ходатайством отсутствуют.
При этом, по мнению экспертизы, не представляется возможным уточнить совокупность признаков таким образом, чтобы она характеризовала комбинированное лекарственное средство Д***, представленное в РУ, поскольку в формуле изобретения RU*** не запатентована возможность использования комбинации двух активных агентов в составе одного лекарственного средства.
2.3.5. Рассмотрение возможности выдачи дополнительного патента на таблетку, охарактеризованную профилем высвобождения
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков
1. Твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая:
(a) соединение двойного действия в количестве от приблизительно 4 до приблизительно 90% в расчете на массу композиции; и
(b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 40, 50, 100, 200 или 400 мг в одной стандартной лекарственной форме; и
где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 10 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
2. Твердая пероральная лекарственная форма по п. 1, где указанная пероральная дозированная форма включает полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] в количестве от 4 до 60% в расчете на массу композиции.
3. Твердая пероральная лекарственная форма по п. 1, где указанная твердая пероральная лекарственная форма представляет собой таблетку.
4. Твердая пероральная лекарственная форма по п. 3, где указанная таблетка представляет собой состав немедленного высвобождения.
5. Твердая пероральная лекарственная форма по п. 4, где указанная таблетка представляет собой ротационно спрессованную таблетку.
6. Твердая пероральная лекарственная форма по п. 1, где фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, Ca или Al, (v) безводный коллоидный диоксид кремния и (vi) тальк.
7. Твердая пероральная лекарственная форма по п. 6, где стеарат Mg используют в количестве от 1,0 мас.% до 6 мас.%, безводный коллоидный диоксид кремния используют в количестве от 0,1 мас.% до 2 мас.%, микрокристаллическая целлюлоза присутствует в количестве от 10 мас.% до 30 мас.%, и кросповидон присутствует в количестве от 1 мас.% до 20 мас.%.
8. Способ получения твердой пероральной лекарственной формы по п. 1 или 2, включающий стадии:
(a) смешивания полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], по крайней мере, с одним фармацевтически приемлемым эксципиентом с получением смеси;
(b) прямого прессования указанной смеси в твердую пероральную лекарственную форму.
9. Способ получения твердой пероральной лекарственной формы по п. 1 или 2, включающий стадии:
(a) смешивания полупентагидрата тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты], по крайней мере, с одним фармацевтически приемлемым эксципиентом с получением смеси;
(b) уплотнения указанной смеси ротационным прессованием;
(c) необязательного смешивания с дополнительными фармацевтически приемлемыми эксципиентами; и
(d) необязательного прессования конечной смеси в твердую пероральную лекарственную форму.
10. Способ получения твердой пероральной лекарственной формы по п. 9, включающий стадии:
(a) просеивания соединения двойного действия и фармацевтически приемлемых ингредиентов с получением просеянного материала;
(b) смешивания просеянного материала с получением смешанного материала;
(c) уплотнения смешанного материала, например, методом ротационного прессования, с получением уплотненного материала;
(d) измельчения уплотненного материала с получением измельченного материала, называемого гранулятом;
(e) необязательного смешивания измельченного материала с дополнительными фармацевтически приемлемыми эксципиентами с получением конечной смеси;
(f) необязательного прессования конечной смеси с получением таблетки; и
(g) необязательного нанесения пленочного покрытия с получением таблетки с пленочным покрытием.
11. Твердая пероральная лекарственная форма, полученная способом по любому из пп. 8 - 10, которая представлена в форме таблетки.
12. Твердая пероральная лекарственная форма по п. 1 или 2, где указанная твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем по меньшей мере приблизительно 40 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
13. Твердая пероральная лекарственная форма по любому из пп. 1 или 2, где (i) соединение двойного действия присутствует в количестве приблизительно 100 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или
(ii) соединение двойного действия присутствует в количестве приблизительно 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или
(iii) соединение двойного действия присутствует в количестве приблизительно 400 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 40 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 70 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 90 мас.% валсартана в форме свободной кислоты.
14. Твердая пероральная лекарственная форма по п. 1 или 2, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч и/или после введения однократной дозы указанной лекарственной формы нормированное относительно дозы среднее содержание в плазме (AUC0-24) составляет от 230 до 400 нгч/мл/мг-экв.
15. Применение твердой пероральной лекарственной формы по п. 14 для повышения скорости абсорбции и/или содержания валсартана в форме свободной кислоты в плазме, причем указанный способ заключается во введении указанной твердой пероральной лекарственной формы пациенту, нуждающемуся в этом.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимом пункте 1 и зависимых пп. 2 - 7 и 12 - 14 формулы изобретения указанного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, Дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, содержащим пояснения патентообладателя на 5 л.;
2) Нотариально заверенная копия Регистрационного удостоверения лекарственного препарата И*** (МНН - валсартан + сакубитрил) N ЛП-000001 от 01.01.2001 - далее РУ на 1 л;
3) Копия Инструкции по медицинскому применению лекарственного препарата И*** (Министерства здравоохранения и социального развития РФ) - далее Инструкция на 17 л;
4) Формула патента РФ *** на 2 л;
5) Данные перечня МНН ВОЗ (Vol. 28, N 1, 2014) на 2л;
6) Выдержки из регистрационного досье препарата "И***", поданного в Министерство Здравоохранения Российской Федерации на 1 л;
7) Формула дополнительного патента на 2 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков.
1. Твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая: (a) соединение двойного действия в количестве от приблизительно 4 до приблизительно 90% в расчете на массу композиции; и (b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 40, 50, 100, 200 или 400 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 10 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
2. Твердая пероральная лекарственная форма по п. 1, где указанная пероральная дозированная форма включает полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] в количестве от 4 до 60% в расчете на массу композиции.
3. Твердая пероральная лекарственная форма по п. 1, где указанная твердая пероральная лекарственная форма представляет собой таблетку.
4. Твердая пероральная лекарственная форма по п. 3, где указанная таблетка представляет собой состав немедленного высвобождения.
5. Твердая пероральная лекарственная форма по п. 4, где указанная таблетка представляет собой ротационно спрессованную таблетку.
6. Твердая пероральная лекарственная форма по п. 1, где фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, Ca или Al, (v) безводный коллоидный диоксид кремния и (vi) тальк.
7. Твердая пероральная лекарственная форма по п. 6, где стеарат Mg используют в количестве от 1,0 мас.% до 6 мас.%, безводный коллоидный диоксид кремния используют в количестве от 0,1 мас.% до 2 мас.%, микрокристаллическая целлюлоза присутствует в количестве от 10 мас.% до 30 мас.%, и кросповидон присутствует в количестве от 1 мас.% до 20 мас.%.
8. Твердая пероральная лекарственная форма по п. 1 или 2, где указанная твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем по меньшей мере приблизительно 40 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
9. Твердая пероральная лекарственная форма по любому из пп. 1 или 2, где (i) соединение двойного действия присутствует в количестве приблизительно 100 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или (ii) соединение двойного действия присутствует в количестве приблизительно 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или (iii) соединение двойного действия присутствует в количестве приблизительно 400 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 40 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 70 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 90 мас.% валсартана в форме свободной кислоты.
10. Твердая пероральная лекарственная форма по п. 1 или 2, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч.
Представленная формула дополнительного патента составлена патентообладателем на основании формулы патента RU***. Пункты 1 и 2 - 7 формулы дополнительного патента идентичны пп. 1 - 7 запатентованной формулы RU***. Зависимые пп. 8 и 9 дополнительного патента идентичны зависимым пп. 12 - 13, а п. 10 составлена на основании зависимого п. 14 указанного патента.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству И***, патентообладатель представляет Регистрационное удостоверение N ЛП-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет. Следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата И***, указанная в представленном разрешении ЛП-000001 от 01.01.2001 с учетом приложенной Инструкции включает следующие признаки:
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: Валсартан + Сакубитрил.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): сакубитрила и валсартана гидратный комплекс натриевых солей 56.551/113.103/226.206 мг [в пересчете на кислотную форму безводную 50/100/200 мг [эквивалентно сакубитрилу 24.3/48.6/97.2 мг, валсартану 25.7/51.4/102.8 мг]], вспомогательные вещества (целлюлоза микрокристаллическая 91.449/34.897/69.794 мг, гипролоза 25.000/25.000/50.000 мг, кросповидон 18.000/18.000/36.000 мг, магния стеарат 6.000/6.000/12.000 мг, тальк 2.000/2.000/4.000 мг, диоксид коллоидный 1.000/1.000/2.000 мг, оболочка - Премикс оболочка белый [гипромеллоза 5.681/5.521/8.422 мг, титана диоксид (E171) 1.138/1.106/1.687 мг, макрогол 400 0.569/0.553/0.843 мг, тальк 0.569/0.553/0.843 мг] 7.957/7.732/11.796 мг, Премикс оболочки желтый [гипромеллоза 0/0.183/0 мг, краситель железа оксид желтый (E172) 0/0.037/0 мг, макрогол 4000 0/0.018/0 мг, тальк 0/0.018/0 мг] 0/0.256/0 мг, Премикс оболочки красный [гипромеллоза 0.014/0.009/0.120 мг, краситель железа оксид красный (E172) 0.003/0.002/0.024 мг, макрогол 4000 0.001/0.001/0.012 мг] 0.019/0.012/0.168 мг, Премикс оболочки черный [гипромеллоза 0.017/0.026 мг, краситель железа оксид черный (E172) 0.003/0/0.005 мг, макрого 4000 0.002/0/0.003 мг, тальк 0.002/0/0.003 мг] 0.024/0/0.036 мг)
Форма выпуска: таблетки, покрытые пленочной оболочкой, 50 мг (25.7 мг + 24.3 мг), 100 мг (51.4 мг+ 48.6 мг), 200 мг (102.8 мг + 97.2 мг) (блистер) 14 x 2 (пачка картонная); таблетки, покрытые пленочной оболочкой, 100 мг (51.4 мг + 48.6 мг), 200 мг (102.8 мг + 97.2 мг) (блистер) 14 x 4 (пачка картонная)
Назначение: Хроническая сердечная недостаточность (II - IV класса по классификации NYHA) у пациентов с систолической дисфункцией с целью снижения риска сердечно-сосудистой смертности и госпитализации по поводу сердечной недостаточности (см. Инструкция с. 7)
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение.
Формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Сравнение характеристик композиции по п. 1 формулы дополнительного патента и характеристики композиции лекарственного средства Интресто показывает следующее.
В представленных РУ (см, МНН, состав лекарственного средства, Форма выпуска), с учетом Инструкции (особенно разделы с. 5 "Всасывание", с. 7 "Показания к применению"), перечня МНН ВОЗ, а также представленного Профиля растворения валсартана (документ 6) охарактеризована твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая: (a) соединение двойного действия; и (b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 50, 100, 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 90 до приблизительно 1 00 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
Учитывая то, что совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, должна быть идентична характеристике композиции лекарственного средства, указанной в разрешении, то обращаем внимание на следующее.
Независимый п. 1 содержит признаки, характеризующие количественное содержание соединения двойного действия выраженные в широком диапазоне, а именно: "соединение двойного действия в количестве от приблизительно 4 до приблизительно 90% в расчете на массу композиции", а также содержит признаки, характеризующие in vitro высвобождение вальсартана, также в широком диапазоне, а именно: "высвобождение в среднем от приблизительно 10 до приблизительно 100 мас.% вальсартана". Следует отметить, что согласно РУ твердая лекарственная форма содержит сакубитрила и вальсарана гидратный комплекс натриевых солей (т.е. соединение двойного действия, которым является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2{"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] по п. 1) в трех конкретных точечных значениях, а именно 50 мг, 100 мг, 200 мг, следовательно, признаки характеризующие содержание данного комплекса в расчете на массу композиции в % не может быть охарактеризован в формуле дополнительного патента в широком диапазоне (от приблизительно 4 до приблизительно 90%). Патентообладателю предлагается уточнить данное значение на основании РУ (раздел "Состав лекарственного средства") и включить полученные конкретные значения в форму.
Также отмечаем то, что согласно представленной "Выдержки из регистрационного досье препарата "И***", поданного в Министерство Здравоохранения Российской Федерации", содержащей график "Профиль растворения вальсартана" из таблеток LCZ696 50 мг, 100 мг, 200 мг следует, что in vitro, что через 30 мин высвобождается в среднем, по мнению экспертизы, от приблизительно 90 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, а не в том широком диапазоне, как это охарактеризовано в п. 1 формулы дополнительного патента, на что указывает и сам паптентообладатель в своих комментариях на с. 4 абзац 3. Патентообладателю предлагается уточнить данное значение на основании указанного документа и включить полученный интервал значений в форму дополнительного патента.
Кроме того патентообладателю, при корректировки независимого п. 1 следует учесть то, что представленное РУ содержит характеристику лекарственного средства Интресто, содержащего сакубитрила и вальсартана гидратный комплекс натриевых солей в количестве 56.551/113.103/226.206 мг, что составляет в пересчете на кислотную форму безводную 50/100/200 мг. В то время, как содержание указанного комплекса в лекарственном препарате Интресто в количестве 40 и 400 мг не охарактеризовано. Таким образом, в данной части характеристика композиции, указанная в формуле изобретения, не идентична характеристике композиции лекарственного средства, на применение которого получено разрешение ЛП-00001 от 01.01.2001. Патентообладателю предлагается уточнить свои притязания в данной части путем исключения количеств 40 и 400 мг из формулы дополнительного патента.
В связи с вышеизложенным, на момент составления настоящего заключения, нельзя признать, что независимый п. 1 формулы дополнительного патента характеризует продукт, который относится к лекарственному средству И***, на применение которого получено разрешение РУ ЛП-000001 от 01.01.2001 в части количественного содержания в фармацевтической композиции активных веществ (сакубитрила и вальсартана гидратный комплекс натриевых солей) 40 и 400 мг, а также in vitro профиля высвобождения вальсартана, и количества комплекса активных веществ в расчете на массу композиции, выраженного в %.
Что касается зависимых пп. 2 - 10 формулы дополнительного патента то отмечаем следующее.
Признаки зависимого п. 2, характеризуют широкий диапазон содержания в лекарственной форме полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты]. Как было указано выше (см. доводы в отношении независимого п. 1) в РУ охарактеризовано содержание данного комплекса только в трех количествах, а не виде широкого диапазона. Патентообладатель вправе уточнить данные количества содержания комплекса в таблетке, выраженное в % и включить данные значения в формулу дополнительного патента, или исключить данный зависимый пункт.
Признаки зависимого п. 3, характеризующие то, что твердая пероральная лекарственная форма представляет собой таблетку, охарактеризовано в РУ (Форма выпуска).
Признаки зависимого п. 4, характеризующие то, что таблетка представляет собой состав немедленного высвобождения, охарактеризованы в Инструкции (с. 5 Фармакокинетика).
Признаки зависимого п. 5, характеризующие то, что таблетка представляет собой ротационно спрессованную таблетку не охарактеризованы в представленных патентообладателем документах. Патентообладатель вправе или представить документы характеризующие то, что таблетка представляет собой ротационно спрессованную таблетку, или исключить данный зависимый пункт из формулы дополнительного патента.
Признаки зависимого п. 6, характеризующие то, что фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, (v) безводный коллоидный диоксид кремния и (vi) тальк, охарактеризованы в РУ (Состав лекарственного средства). При этом следует отметить, что в РУ не охарактеризовано то, что в качестве фармацевтичски приемлемых эксцепиентов могут быть использованы стеарат Ca или Al. Патентообладатель вправе исключить указанные эксцепиенты из формулы дополнительного патента, поскольку в данной части формула не идентична характеристике лекарственного средства И***.
Что касается признаков зависимого п. 7, то необходимо отметить, что он содержит характеристику количественного содержания эксцепиентов в таблетке в широком диапазоне. Как было указано выше (см. доводы экспертизы в отношении независимого п. 1), нельзя признать, что такая формулировка признаков позволит признать то, что формула изобретения в данной части идентична характеристике лекарственного средства И***. Патентообладатель вправе уточнить данные значения на основании РУ или исключить их из формулы дополнительного патента.
Что касается признаков зависимого п. 8 отмечаем то, что, по мнению экспертизы с учетом представленного профиля растворения вальсартана (см. Выдержки из регистрационного досье препарата "И***", поданного в Министерство Здравоохранения Российской Федерации на 1 л;) твердая пероральная лекарственная форма характеризуется профилем растворения in vitro, таким что через 10 мин высвобождается в среднем по меньшей мере приблизительно 50 - 60 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли (см. графики охарактеризованных в РУ таблеток: 50, 100, 200 мг), а не приблизительно 40 мас.%, как это указано в п. 8. Патентообладатель вправе представить данные указывающие на то, что твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем по меньшей мере приблизительно 40 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, или исключить данный пункт из формулы дополнительного патента.
Что касается зависимого п. 9 то необходимо отметить, следующее. Согласно данному зависимому пункту охарактеризован профиль высвобождения вальсартана из таблетки, при этом таблетка содержит соединение двойного действия (сакубитрила и валсартана гидратный комплекс натриевых солей) в количестве 100 мг, 200 мг, 400 мг. Признаки п. 9, характеризующие профиль высвобождения вальсартана из таблеток содержащих 100 и 200 мг, охарактеризованы в Выдержке из регистрационного досье препарата "Интресто", поданного в Министерство Здравоохранения Российской Федерации (график Профиль растворения вальсартана). Что касается признаков характеризующие профиль высвобождения вальсартана из таблеток, содержащих соединение двойного действия в количестве 400 мг то, следует отметить, что согласно РУ зарегистрированы только таблетки содержащие соединение двойного действия в количестве 100 и 200 мг. Таким образом, зависимый п. 9 в части профиля высобождения вальсартана из таблетки содержащей соединение двойного действия в количестве 400 мг не идентичен зарегистрированному лекарственному средству И***. Патентообладатель вправе исключить характеристику высвобождения таблетки содержащей соединение двойного действия в количестве 400 мг из формулы дополнительного патента.
Признаки зависимого п. 10, характеризующие то, что твердая пероральная лекарственная форма, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч, охарактеризованы в Инструкции (с. 5 Фармакокинетика. Всасывание).
Таким образом, представленная патентообладателем формула дополнительного патента, не характеризует продукт, который относится к лекарственному средству И*** на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, не идентична характеристике композиции лекарственного средства, указанного в разрешении.
С учетом вышеизложенного основания для выдачи дополнительного патента с формулой, представленной заявителем совместно с ходатайством отсутствуют. Патентообладателю был направлен запрос с указанным выводом экспертизы.
В ответ на запрос патентообладатель представил скорректированную формулу дополнительного патента.
1. Твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая: (a) соединение двойного действия в количестве от 28 до 60% в расчете на массу композиции; и (b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 50, 100 или 200 мг в одной стандартной лекарственной форме; и где твердая
пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 90 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
2. Твердая пероральная лекарственная форма по п. 1, где указанная твердая пероральная лекарственная форма представляет собой таблетку.
3. Твердая пероральная лекарственная форма по п. 2, где указанная таблетка представляет собой состав немедленного высвобождения.
4. Твердая пероральная лекарственная форма по п. 3, где указанная таблетка представляет собой ротационно спрессованную таблетку.
5. Твердая пероральная лекарственная форма по п. 1, где фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, (v) безводный коллоидный диоксид кремния и (vi) тальк.
6. Твердая пероральная лекарственная форма по п. 6, где стеарат Mg используют в количестве от 1,0 мас.% до 6 мас.%, безводный коллоидный диоксид кремния используют в количестве от 0,1 мас.% до 2 мас.%, микрокристаллическая целлюлоза присутствует в количестве от 10 мас.% до 30 мас.%, и кросповидон присутствует в количестве от 1 мас.% до 20 мас.%.
7. Твердая пероральная лекарственная форма по п. 1, где указанная твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем 50 - 60 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
8. Твердая пероральная лекарственная форма по любому из п. 1, где (i) соединение двойного действия присутствует в количестве приблизительно 100 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты, или (ii) соединение двойного действия присутствует в количестве приблизительно 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 10 мин высвобождается в среднем приблизительно 50 мас.% валсартана в форме свободной кислоты, через 20 мин высвобождается в среднем приблизительно 85 мас.% валсартана в форме свободной кислоты, через 30 мин высвобождается в среднем приблизительно 95 мас.% валсартана в форме свободной кислоты.
9. Твердая пероральная лекарственная форма по п. 1, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч.
Представленная формула дополнительного патента составлена патентообладателем на основании формулы патента RU***.
Сравнение характеристик композиции по п. 1 формулы дополнительного патента и характеристики композиции лекарственного средства Интресто с учетом ответов заявителя и скорректированной формулы дополнительного патента, показывает следующее.
В своем ответе патентообладатель указывает на то, что содержание соединения двойного действия в расчете на массу композиции для таблеток 50 мг (масса таблетки 200 мг), 100 мг (масса таблетки 200 мг) и 200 мг (масса таблетки 400 мг), равно, соответственно, 28,3%, 56,6% и 56,6%. Также в данном ответе представлены дополнительные сведения, раскрывающие то, что зарегистрированная таблетка представляет собой ротационно-спресованную таблетку. Данным документом является: Выдержка из регистрационного досье препарата "И***" (Приложение 3).
Также в указанном ответе на с. 1, патентообладатель отмечает, что согласно РУ твердая лекарственная форма содержит сакубитрила и вальсарана гидратный комплекс натриевых солей (т.е. соединение двойного действия, которым является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] по п. 1) в трех конкретных точечных значениях, а именно 50 мг, 100 мг, 200 мг, что соответствует 28,2%, 56,6% и 56,6%.
Таким образом в представленных РУ (см, МНН, состав лекарственного средства, Форма выпуска), с учетом Инструкции (особенно разделы с. 5 "Всасывание", с. 7 "Показания к применению"), перечня МНН ВОЗ, а также представленного Профиля растворения валсартана (документ 6) охарактеризована твердая пероральная лекарственная форма, предназначенная для доставки терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, включающая: (a) соединение двойного действия в количестве от 28 до 60% в расчете на массу композиции; и (b) по крайней мере, один фармацевтически приемлемый эксципиент, где соединением двойного действия является полупентагидрат тринатриевой соли [3-((1S,3R)-1-бифенил-4-илметил-3-этоксикарбонил-1-бутилкарбамоил)пропионат-(S)-3'-метил-2'-(пентаноил{2"-(тетразол-5-илат)бифенил-4'-илметил}амино)масляной кислоты] и где соединение двойного действия присутствует в количестве 50, 100, 200 мг в одной стандартной лекарственной форме; и где твердая пероральная лекарственная форма характеризуется таким профилем растворения in vitro, что через 30 мин высвобождается в среднем от приблизительно 90 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
Согласно представленной "Выдержки из регистрационного досье препарата "И***", поданного в Министерство Здравоохранения Российской Федерации", содержащей график "Профиль растворения вальсартана" из таблеток LCZ696 50 мг, 100 мг, 200 мг следует, что in vitro, что через 30 мин высвобождается в среднем, от приблизительно 90 до приблизительно 100 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли.
Таким образом независимый п. 1 формулы дополнительного патента характеризует продукт, который относится к лекарственному средству И***, на применение которого получено разрешение РУ ЛП-0000001 от 01.01.2001.
Что касается зависимых пп. 2 - 9 формулы дополнительного патента то необходимо отметить следующее.
Признаки зависимого п. 2, характеризующие то, что твердая пероральная лекарственная форма представляет собой таблетку, охарактеризованы в РУ (Форма выпуска).
Признаки зависимого п. 3, характеризующие то, что таблетка представляет собой состав немедленного высвобождения, охарактеризованы в Инструкции (с. 5 Фармакокинетика).
Признаки зависимого п. 4, характеризующие то, что таблетка представляет собой ротационно спрессованную таблетку охарактеризованы в выдержке из регистрационного досье в представленном патентообладателем.
Признаки зависимого п. 5, характеризующие то, что фармацевтически приемлемые эксципиенты включают (i) микрокристаллическую целлюлозу, (ii) гидроксипропилцеллюлозу, (iii) кросповидон, (iv) стерат Mg, (v) безводный коллоидный диоксид кремния и (vi) тальк, охарактеризованы в РУ (раздел "Состав лекарственного средства").
Признаков зависимого п. 6, характеризующие то, стеарат Mg используют в количестве от 1,0 мас.% до 6 мас.%, безводный коллоидный диоксид кремния используют в количестве от 0,1 мас.% до 2 мас.%, микрокристаллическая целлюлоза присутствует в количестве от 10 мас.% до 30 мас.%, и кросповидон присутствует в количестве от 1 мас.% до 20 мас.%, охарактеризованы в РУ (раздел "Состав лекарственного средства").
В отношении признаков зависимого п. 7 отмечаем то, что с учетом представленного профиля растворения вальсартана (см. Выдержки из регистрационного досье препарата "Интресто", поданного в Министерство Здравоохранения Российской Федерации на 1 л;) твердая пероральная лекарственная форма характеризуется профилем растворения in vitro, таким что через 10 мин высвобождается в среднем по меньшей мере приблизительно 50 - 60 мас.% валсартана в форме свободной кислоты или его фармацевтически приемлемой соли (см. графики охарактеризованных в РУ таблеток: 50, 100, 200 мг).
Признаки п. 8, характеризующие профиль высвобождения вальсартана из таблеток содержащих 100 и 200 мг, охарактеризованы в Выдержке из регистрационного досье препарата "И***", поданного в Министерство Здравоохранения Российской Федерации (график Профиль растворения вальсартана).
Признаки зависимого п. 9, характеризующие то, что твердая пероральная лекарственная форма, предназначенная для доставки, после введения, терапевтически эффективного количества валсартана в форме свободной кислоты или его фармацевтически приемлемой соли, причем после введения однократной дозы указанной лекарственной формы скорость абсорбции валсартана в форме свободной кислоты tмакс составляет от 1 до 2,2 ч, охарактеризованы в Инструкции (с.5 Фармакокинетика. Всасывание).
Таким образом, представленная патентообладателем формула дополнительного патента, характеризует продукт, который относится к лекарственному средству И*** на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, указанного в разрешении.
На основании изложенного считаем возможным выдать дополнительный патент с формулой, представленной патентообладателем в дополнительных материалах.
2.3.6. Рассмотрение возможности выдачи дополнительного патента на фармацевтическую композицию, таблетку и способ ее изготовления
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Фармацевтическая композиция для лечения гиперфосфатемии, включающая оксигидроксид железа в количестве от 10 до 80% (по весу), по отношению к общему весу композиции, который присутствует в количестве более 300 мг на лекарственную форму, изготовленная в виде лекарственных форм, приспособленных к распаду в ротовой полости или в малом количестве жидкости перед проглатыванием.
2. Фармацевтическая композиция по п. 1, отличающаяся тем, что оксигидроксид железа присутствует в количестве около 800 мг на лекарственную форму.
3. Способ приготовления таблеток, содержащих композицию по п. 1, путем прямого прессования или сухого гранулирования.
4. Таблетка для лечения гиперфосфатемии, полученная способом по п. 3.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в пунктах 1 - 4 формулы формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, Дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) заявление на 2 л.;
2) библиографические данные и формула патента RU*** C2 на 4 л.;
3) Копия Регистрационного удостоверения (РУ) N ЛП-00001 от 01.01.2001 на 2 л;
4) Скорректированная формула изобретения для дополнительного патента на 4 л.
Согласно п. 2 ст. 1363 Кодекса продление срока действия исключительного права на изобретение осуществляется в отношении изобретения, относящегося к лекарственному средству, пестициду или агрохимикату.
Согласно п. 2 ст 1363 Кодекса при продлении срока действия исключительного права выдается дополнительный патент с формулой, содержащей совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков.
1. Фармацевтическая композиция для лечения гиперфосфатемии, включающая оксигидроксид железа в количестве от 10 до 80% (по весу), по отношению к общему весу композиции, который присутствует в количестве более 300 мг на лекарственную форму, изготовленная в виде лекарственных форм, приспособленных к распаду в ротовой полости или в малом количестве жидкости перед проглатыванием.
2. Фармацевтическая композиция по п. 1, отличающаяся тем, что оксигидроксид железа присутствует в количестве около 800 мг на лекарственную форму.
3. Способ приготовления таблеток, содержащих композицию по п. 1, путем прямого прессования или сухого гранулирования.
4. Таблетка для лечения гиперфосфатемии, полученная способом по п. 3.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству В***, патентообладатель представляет Регистрационное удостоверение N ЛП-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата В***, указанная в представленном разрешении ЛП-000001 от 01.01.2001 включает следующие признаки:
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: Комплекс железа оксигидроксида, сахарозы и крахмала.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): комплекс железа оксигидроксида, сахарозы и крахмала 2500.00 мг, [эквивалент железа 500 мг], вспомогательные вещества (ароматизатор "лесная ягода" 40.00 мг, неогесперидина дигидрохалкон 0.01 мг, магния стеарат 25.00 мг, кремния диоксид коллоидный безводный 12.49 мг).
Форма выпуска: таблетки жевательные, 500 мг (блистер) 6 x 5 (пачка картонная); таблетки жевательные, 500 мг (флакон) 30/90 x 1 (пачка картонная).
1) Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Для возможности сравнения композиции по независимому пункту 1 дополнительного патента и композиции лекарственного средства В*** патентообладателем представлено только вышеуказанное РУ. Однако данное РУ не содержит достаточно сведений необходимых для проведения указанного сравнения, поскольку в РУ не указано назначение лекарственного средства В***. Согласно независимому п. 1, назначением фармацевтической композиции является лечением гиперфосфатемии.
Данные сведения могут быть отражены в Инструкции по медицинскому применения лекарственного средства В***, которую патентообладатель вправе представить для возможности проведения сравнения композиции по независимым пунктам дополнительного патента и композиции лекарственного средства В***.
Притязания патентообладателя по п. 1 формулы дополнительного патента содержат признаки, характеризующие количественный состав активного вещества в композиции, выраженные в виде диапазона процентного содержания по весу оксигидроксид железа, по отношению к общему весу композиции. При этом, также в п. 1 указано, что оксигидроксид железа присутствует в количестве более 300 мг на лекарственную форму, что также представляет собой диапазон количества вещества в лекарственной форме.
Учитывая то, что совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, должна быть идентична характеристике композиции лекарственного средства, указанной в разрешении, то отмечаем, что согласно РУ (см, раздел "Состав лекарственного средства") количественное содержание оксигидроксид железа в лекарственном средстве В*** охарактеризовано конкретным количеством, а не диапазоном. Патентообладателю предлагается уточнить конкретное значения содержания оксигидроксид железа, на основании состава лекарственного средства В***, указанного в РУ. Также предлагается включить данное значение в формулу дополнительного патента, поскольку только в этом случае можно будет признать, что совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, будет идентична характеристике композиции лекарственного средства, указанной в разрешении.
Патентообладатель вправе скорректировать п. 1, путем уточнения количества вещества, таким образом, чтобы характеристика запатентованной композиции была идентична композиции лекарственного средства на применение которого получено РУ ЛП-000001 от 01.01.2001.
Таким образом, на момент составления настоящего заключения, нельзя признать, что п. 1 формулы дополнительного патента характеризуют продукт, который относится к лекарственному средству В***, на применение которого получено разрешение РУ ЛП-000001 от 01.01.2001.
2) Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение.
Независимый пункт формулы дополнительного патента, характеризующий "Способ" по п. 3 согласно указанному родовым понятиям не относятся к объекту изобретения - продукт.
Т.е. представленная патентообладателем формула дополнительного патента в части п. 3 не характеризует продукт, который относится к лекарственному средству В*** на применение которого получено разрешение, поскольку родовое понятие указанных пунктов не относится к объекту изобретения продукт.
В связи с этим ходатайство патентообладателя в части данных пунктов не может быть удовлетворено, поскольку объект по п. 3, не характеризуют продукт как таковой.
3) Что касается независимого п. 4, характеризующего объект "Таблетка", отмечаем следующее.
Несмотря на то, что родовое понятие данного объекта, характеризует продукт как таковой, однако нельзя признать, что независимый п. 3 содержит совокупность признаков, характеризующую продукт, на применение которого получено разрешение.
Таблетка по п. 4 не содержит совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентичную характеристике композиции лекарственного средства, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Таким образом, нельзя признать, что п. 4 формулы представленной патентообладателем для выдачи дополнительного патента характеризует продукт, который относится к лекарственному средству В***, на применение которого получено разрешение РУ ЛП-000001 от 01.01.2001, а следовательно, формула дополнительного патента не характеризует продукт, который относится к лекарственному средству В*** на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, не идентична характеристике композиции лекарственного средства, указанного в разрешении.
С учетом вышеизложенного основания для выдачи дополнительного патента с формулой, представленной заявителем совместно с ходатайством отсутствуют.
Патентообладатель вправе представить дополнительные данные, например, Инструкцию по медицинскому применению и/или любую другую информацию, характеризующую лекарственное средство В***, а также скорректированную формулу дополнительного патента, составленную с учетом замечаний экспертизы и требования подпункта 1 и 2 пункта 8 Порядка.
В ответ на данный запрос патентообладатель представил скорректированную формулу дополнительного патента, где исключил объекты "Применение... для лечения", "Способ" и "Таблетка", а также представил запрашиваемую экспертизой Инструкцию по применению лекарственного препарата для медицинского применения В*** на 11 л в 1 экз (далее - Инструкция), где раскрыто назначение заявленной композиции, которым является лечения гиперфосфатемии,
1. Фармацевтическая композиция для лечения гиперфосфатемии, включающая оксигидроксид железа (эквивалент железа 500 мг на лекарственную форму), сахарозу и крахмал, изготовленная в виде жевательной таблетки.
В представленной формулы необходимо отметить следующее.
Из пояснений патентообладателя следует, что независимый п. 1 представленной формулы дополнительного патента им уточнен на основании независимого п. 1 формулы патента RU***. Однако, ни в одном из указанных пунктов запатентованной формулы не охарактеризовано то, что фармацевтическая композиция включает "эквивалент железа 500 мг на лекарственную форму". В независимом п. 1 патента RU*** охарактеризовано количественное содержание оксигидроксида железа в лекарственной форме, при этом отсутствуют признаки, характеризующие эквивалентное содержание железа в лекарственной форме как таковые. Следует отметить, что в формуле патента RU*** зависимый п. 2 охарактеризовано то, что оксигидроксид железа присутствует в количестве около 800 мг на лекарственную форму.
Таким образом, нельзя признать, что независимый п. 1 формулы дополнительного патента уточнен на основании формулы патента RU***. Экспертиза принимает во внимание представленный патентообладателем в ответе расчет содержания железа в оксигидроксиде железа, входящим в состав лекарственной формы, основанный на описании патента и отмечает следующее. Данный расчет, позволяет уточнить количественное содержание бета оксигидроксиде железа по независимому п. 1 дополнительного патента на основании признаков п. 2 RU*** а именно: "оксигидроксид железа присутствует в количестве 800 мг на лекарственную форму", что соответствует представленному РУ.
Дополнительный патент может быть выдан на следующую совокупность признаков:
1. Фармацевтическая композиция для лечения гиперфосфатемии, включающая оксигидроксид железа в количестве 800мг на лекарственную форму, сахарозу и крахмал, изготовленная в виде жевательной таблетки.
2.3.7. Рассмотрение возможности выдачи дополнительного патента на набор.
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Набор для лечения рака у млекопитающего, содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль, в дозировках, подходящих для введения дважды в день, а также необязательно содержащий противоопухолевый агент, выбранный из группы, включающей в себя цисплатин, 1,3-бис(2-хлорэтил)-1-нитрозомочевину, доцетаксел, доксорубицин, эпирубицин, этопозид, метотрексат, митомицин, гемцитабин, карбоплатин, гефитиниб, пеметрексед, авастин, цетуксимаб и паклитаксел, а также содержащий памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента.
Заявитель просит продлить срок действия исключительного права на изобретение, описанное в патенте RU***, в отношении независимого п. 1 относящегося к объекту "Набор".
К ходатайству патентообладатель представляет следующие документы:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** и пояснения патентообладателя на 2 л.
2) Копия первого Регистрационного Удостоверения ЛП-000001 от 01.01.2001, содержащего сведения о регистрационном номере и дате получения первого разрешения уполномоченного органа на применение продукта Т*** (далее - РУ);
3) Копия титульного листа и формулы патента RU*** на 4 л;
4) Приложение 1 и Приложение 2, содержащие сведения о структуре и названиях Гимерацила и Калия оксоната, соответственно;
5) Копия Инструкции по применяю лекарственного препарата для медицинского применения Т*** (далее - Инструкция) на 31 л.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU***, к лекарственному средству Т*** патентообладатель представляет документы, включающие Регистрационное удостоверение ЛП-000001 от 01.01.2001. Данное Регистрационное удостоверение выдано впервые со сроком действия 5 лет.
Характеристика лекарственного препарата Т***, указанная в представленном разрешении ЛП-00001 от 01.01.2001 включает следующие признаки:
Международное непатентованное наименование или химическое (группировочное) наименование лекарственного препарата: (Тегафур/Гимерацил/Отерацил.
Лекарственная форма, дозировка(-и): капсулы, 15мг/4.35 мг/11.8 мг, 20мг/5.8 мг/15.8 мг.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): тегафур 15./20.00 мг, гимерацил 4.35/5.80, отерацил калия 14.70/19.60 мг [в пересчете на отерацил 11.80/15.80 мг], вспомогательные вещества (лактозы моногидрат 70.2/93.6 мг, магния стеарат 0.3/0.4 мг) оболочка [корпус - желатин 19.7752/19.7752 мг, титана диоксид 0.8400/0.8400 мг, натрия лаурилсульфат 0.0248/0.0248 мг, тальк в следовых количествах/ в следовых количествах, крышечка - желатин 13.4155/13.1835 мг, титана диоксид 0.2400/0.5600 мг, железа оксид красный 0.0880/0 мг, натрия лаурилсульфат 0.0165/0.0165 мг, тальк в следовых количествах/ в следовых количествах], чернила - железа оксид желтый в следовых количествах/в следовых количествах, железа оксид красный 0/в следовых количествах, FD&C синий N 2 алюминиевый лак (E132) в следовых количествах/в следовых количествах, карнаубский воск в следовых количествах/в следовых количествах, отбеленный шеллак в следовых количествах/в следовых количествах, глицерил моноолеат в следовых количествах/в следовых количествах).
Форма выпуска: капсулы, 15мг/4.35 мг/11.8 мг, 20 мг/5.8 мг/15.8 мг (блистер) 10 x 3/6/9 (пачка картонная).
Назначение: противоопухолевое средство - антиметаболит (см. Инструкцию).
Характеристика композиции описанной в независимом п. 1 формулы изобретения RU*** включает следующие признаки:
Набор для лечения рака у млекопитающего, содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль, в дозировках, подходящих для введения дважды в день, а также необязательно содержащий противоопухолевый агент, выбранный из группы, включающей в себя цисплатин, 1,3-бис(2-хлорэтил)-1-нитрозомочевину, доцетаксел, доксорубицин, эпирубицин, этопозид, метотрексат, митомицин, гемцитабин, карбоплатин, гефитиниб, пеметрексед, авастин, цетуксимаб и паклитаксел, а также содержащий памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента.
Сравнение характеристик композиции, запатентованной в независимом п. 1 и активного ингредиента лекарственного средства Тейсуно показывает следующее.
Признак п. 1, характеризующие набор для лечения рака содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин (гимерацил), оксоновую кислоту или ее фармацевтически приемлемую соль (а именно, отерацил калия), охарактеризованы РУ см. Разделы: Лекарственная форма, Состав лекарственного средства, Форма выпуска, с учетом Приложения 1 и 2. Признаки, характеризующие дозировки (используемых активных веществ), подходящие для введения дважды в день, а также то, что набор содержит памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента, охарактеризованы в Инструкции по применению лекарственного препарата для медицинского применения Тейсуно в разделе "Способ применения и дозы" (см. с. 9 Инструкции).
Таким образом, совокупность признаков запатентованного изобретения в независимом п. 1 формулы изобретения патента RU*** и характеристики композиции, являющейся активным ингредиентом лекарственного средства Т*** (Тегафур/Гимерацил/Отерацил) на применение которого получено РУ ЛП-00001 от 01.01.2001 совпадают в следующей части:
Набор для лечения рака у млекопитающего, содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль, в дозировках, подходящих для введения дважды в день, а также содержащий памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента.
Согласно п. 2 ст. 1363 Кодекса при продлении срока действия исключительного права выдается дополнительный патент с формулой, содержащей совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
Т.о. патентообладатель вправе представить формулу, содержащую совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
В случае если патентообладатель сохранит в скорректированной формуле признаки, характеризующие то, что набор необязательно содержит противоопухолевый агент, выбранный из группы, включающей в себя цисплатин, 1,3-бис(2-хлорэтил)-1-нитрозомочевину, доцетаксел, доксорубицин, эпирубицин, этопозид, метотрексат, митомицин, гемцитабин, карбоплатин, гефитиниб, пеметрексед, авастин, цетуксимаб и паклитаксел, то в данном случае нельзя будет признать, что характеристики запатентованной композиции по п. 1 совпадают с характеристиками активного ингредиента лекарственного средства, на применение которого получено Регистрационное удостоверение N ЛП-000001 от 01.01.2001
В поступившем ответе патентообладатель не согласен с необходимостью дополнительного ограничения скорректированной формулы в части одного из активных ингредиентов набора, только калиевой солью оксоновой кислоты, как это было предложено экспертизой.
Доводы патентообладателя заключаются в следующем. Международное непатентованное название Отерацил относится к самой оксоновой кислоте, а не к ее соли, в том числе калиевой. Именно поэтому как в Регистрационном Удостоверении (РУ), так и в инструкции для препарата Тейсуно использованы оба эти наименования, а именно Отерацил и Отерацил калия. Более того, свидетельством тому, что Отерацил и Отерацил калия являются разными индивидуальными химическими соединениями (в первом случае это кислота, во втором-соль) является информация о разных количествах действующего вещества в случае, если оно используется в виде кислоты (11,8 мг или 15,8 мл) и в виде соли (14,7 мг и 19,6 мг) (см. Инструкция с. 1, "Действующие вещества"). Для подтверждения вышеуказанного патентообладатель представляет два документа, раскрывающих тот факт, что МНН Отерацил является наименованием оксоновой кислоты, а не ее соли (см. Приложение 1 и 2 к ответу от 29.01.2016).
Экспертиза принимает во внимание представленные дополнительные сведения (см. Приложение 1 и 2), и также согласна с мнением патентообладателя.
Таким образом, повторно рассмотрена возможность продления срока действия исключительного права на изобретение и удостоверяющего его патента RU***, со следующей совокупностью признаков.
Набор для лечения рака у млекопитающего, содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль, в дозировках, подходящих для введения дважды в день, а также содержащий памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента.
Уточненная формула, соответствует требованиям предъявляемым к формуле дополнительного патента, согласно п. 2 ст. 1363 Кодекса по причинам указанным ниже.
Совокупность признаков скорректированной формулы изобретения, характеризующие набор для лечения рака содержащий тегафур, 2,4-дигидрокси-5-хлорпиридин (гимерацил), оксоновую кислоту (Отерацил) или ее фармацевтически приемлемую соль (а именно, отерацил калия), охарактеризованы РУ см. Разделы: МНН, Лекарственная форма, Состав лекарственного средства, Форма выпуска, с учетом Приложения 1 и 2. Признаки, характеризующие дозировки (используемых активных веществ), подходящие для введения дважды в день, а также то, что набор содержит памятку, напоминающую, что указанные тегафур, 2,4-дигидрокси-5-хлорпиридин, оксоновую кислоту или ее фармацевтически приемлемую соль следует принимать в голодном состоянии пациента, охарактеризованы в Инструкции по применению
лекарственного препарата для медицинского применения Тейсуно в разделе "Способ применения и дозы" (см. с. 9 Инструкции).
Таким образом, совокупность признаков представленная патентообладателем для дополнительного патента (см. выше) и характеристики композиции, являющейся активным ингредиентом лекарственного средства Т*** (Тегафур/Гимерацил/Отерацил) на применение которого получено РУ ЛП-000001 от 01.01.2001 совпадают.
На основании изложенного считаем возможным выдать дополнительный патент с формулой, представленной патентообладателем, содержащей совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение РУ ЛП-0000001.
2.3.8. Рассмотрение возможности выдачи дополнительного патента на применение
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Применение экстракта плодов расторопши пятнистой для изготовления гепатопротекторного лекарственного средства.
2. Применение экстракта плодов расторопши пятнистой для лечения тяжелых поражений печени начинается с дозы 1 капсула 3 раза в день ежедневно в течение 10 дней и далее через день по 1 капсуле в течение месяца.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в пунктах 1 - 2 формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, Дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) заявление и приложение, содержащее доводы заявителя на 3 л.;
2) Распечатка с сайта Госреестра лекарственных средств Регистрационного удостоверения ЛП-000001 от 01.01.2001 на 1 л;
3) Инструкция по медицинскому применению лекарственного препарата для медицинского применения К*** (далее - Инструкция) на 17 л;
4) Формула дополнительного патента на 1 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков.
1. Применение экстракта плодов расторопши пятнистой для изготовления гепатопротекторного лекарственного средства.
2. Применение экстракта плодов расторопши пятнистой для лечения тяжелых поражений печени начинается с дозы 1 капсула 3 раза в день ежедневно в течение 10 дней и далее через день по 1 капсуле в течение месяца.
Представленная формула дополнительного патента составлена патентообладателем на основании формулы патента RU***.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству К***, патентообладатель представляет Регистрационное удостоверение N ЛСР-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано со сроком действия 5 лет. Следовательно РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата К***, указанная в представленном разрешении ЛП-00001 от 01.01.2001 с учетом приложенной Инструкции включает следующие признаки:
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: экстракт плодов расторопши пятнистой. Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): Активное вещество: - сухой экстракт плодов расторопши пятнистой 163.6-225 мг (эквивалент 90 мг силимарина), вспомогательные вещества: лактозы моногидрат 38.2 - 7.5 мг, целлюлоза микрокристаллическая (тип 101) 38.2 - 7.5 мг, крахмал пшеничный 15.5 мг, повидон К25 3.7 мг, полисорбат 80 3.7 мг, кремния диоксид коллоидный безводный 3.4 мг, маннитол 80 мг, кросповидон 14 мг, натрия гидрокарбонат 6 мг, магния стеарат 3.7 мг; Состав оболочки капсулы: железа оксид черный 0.02%, железа оксид красный 0.03%, титана диоксид 0.6666%, железа оксид желтый 0.35%, желатин (до 100%
Форма выпуска: Капсулы твердые желатиновы - пачка картонная.
Назначение: гепатопротектор (Инструкция, с. 1).
Далее, согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение.
Формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
1. Сравнение характеристик композиции по п. 1, формулы дополнительного патента и характеристики композиции лекарственного средства К*** показывает следующее.
Согласно независимому п. 1 предложено применение экстракта плодов расторопши пятнистой для изготовления гепатопротекторного лекарственного средства.
В представленных РУ (см, МНН, состав лекарственного средства) и Инструкции (см, "Показания к применению" с. 4) охарактеризовано применение экстракта плодов расторопши пятнистой для изготовления гепатопротекторного лекарственного средства.
Таким образом, представленная патентообладателем формула в части п. 1 дополнительного патента характеризует продукт, который относится к лекарственному средству К*** на применение которого получено разрешение, поскольку в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, указанной в разрешении ЛП-00001 от 01.01.2001.
2. Сравнение характеристик композиции по п. 2, формулы дополнительного патента и характеристики композиции лекарственного средства К*** показывает следующее.
Согласно независимому п. 2 предложено применение экстракта плодов расторопши пятнистой для лечения тяжелых поражений печени начинается с дозы 1 капсула 3 раза в день ежедневно в течение 10 дней и далее через день по 1 капсуле в течение месяца.
В соответствии с п. 1 ст. 1350 ГК РФ в качестве изобретения охраняется техническое решение в любой области, относящееся к продукту (в частности, устройству, веществу, штамму микроорганизма, культуре клеток растений или животных) или способу (процессу осуществления действий над материальным объектом с помощью материальных средств), в том числе к применению продукта или способа по определенному назначению.
Следует отметить, что если изобретение охарактеризованное в п. 1 дополнительного патента относится к применению продукта по определенному назначении, то изобретение по п. 2 относится к применению способа по определенному назначению, поскольку охарактеризовано определенными приемами способа лечения пациента, а именно: "начинается с дозы 1 капсула 3 раза в день ежедневно в течение 10 дней и далее через день по 1 капсуле в течение месяца".
Согласно п. 2 ст. 1363 ГК РФ при продлении срока действия исключительного права выдается дополнительный патент с формулой, содержащей совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
В связи с этим нельзя признать, что п. 2 формулы дополнительного патента, содержит совокупность признаков запатентованного изобретения, характеризующую продукт, на применение которого получено разрешение.
На основании изложенного продление исключительного права возможно исключительно в отношении независимого пункта 1.
2.3.9. Рассмотрение возможности выдачи дополнительного патента на ингалятор
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Мультидозовое ингаляторное устройство для сухого порошка, содержащее аклидиний, откалиброванное для обеспечения введения после его задействования отмеренной номинальной дозы аклидиния, эквивалентной 400 мкг (плюс/минус 10%) мкг аклидиния бромида, причем в ингаляторном устройстве аклидиний является единственным активным ингредиентом.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимом пункте 1 формулы изобретения указанного патента и выдачи дополнительного патента.
Ходатайство рассмотрено с учетом действующего законодательства на момент его подачи, а именно: п. 2 ст. 1363 Гражданского Кодекса РФ (далее - Кодекс), Административный регламент предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патент, дата начала действия: 8 января 2016 г. (далее - Регламент) и Порядок выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение, дата начала действия: 8 января 2016 г. (далее - Порядок).
Патентообладателем представлены соответствующие документы, необходимые для продления срока действия патента в соответствии с пунктом 16 Регламента. Данными документами являются:
1) Заявление о продлении срока действия исключительного права на изобретение по патенту RU*** с приложением, включающим доводы заявителя, на 3 л;
2) Скорректированная формула изобретения для дополнительного патента (с комментариями и без) на 2 л;
3) Копия Регистрационного удостоверения (РУ) N ЛП-000001 от 001.01.2001 на 2 л;
4) Копия Инструкции по применению лекарственного препарата для медицинского применения Б*** (далее - Инструкция) на 18 л.
Патентообладатель просит продлить срок действия исключительного права и выдать ему дополнительный патент со следующей совокупностью признаков:
1. Мультидозовое ингаляторное устройство для сухого порошка, содержащее аклидиний, откалиброванное для обеспечения введения после его задействования отмеренной номинальной дозы аклидиния, эквивалентной 400 мкг (плюс/минус 10%) мкг аклидиния бромида, причем в ингаляторном устройстве аклидиний является единственным активным ингредиентом.
Представленная формула дополнительного патента составлена на основании запатентованной формулы изобретения RU***.
Для возможности отнесения изобретения, охарактеризованного в независимом пункте формулы изобретения, защищенного патентом RU*** к лекарственному средству Б***, патентообладатель представляет Регистрационное удостоверение N ЛП-000001 от 01.01.2001.
Согласно подпункта 2 пункта 8 Порядка, устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения: для лекарственных средств - в соответствии с положением Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выданное со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата.
Представленное РУ выдано впервые со сроком действия 5 лет, следовательно, РУ соответствует требованию подпункта 2 пункта 8 Порядка.
Характеристика лекарственного препарата Д***, указанная в представленном разрешении N ЛП-000001 от 01.01.2001 включает следующие признаки:
Торговое наименование: Б***.
Международное непатентованное, или группировочное, или химическое наименование лекарственного препарата: Аклидиния бромид.
Состав лекарственного средства (качественный и количественный состав действующих и вспомогательных веществ): аклидиния бромид микронизированный 0.400 мг, вспомогательные вещества ( -лактозы моногидрат 12.60 мг).
-лактозы моногидрат 12.60 мг).
Форма выпуска: порошок для ингаляций дозированный, 322 мкг/доза (ингалятор) 30 доз x 1, 60 доз x 1/3 (пачка картонная).
Назначение: м-холиноблокатор. Облегчение симптомов хронической обструктивной болезни легких у взрослых (Инструкция, стр. 1 раздел "Фармакотерапевтическая группа", стр. 7 раздел "Показания к применению").
Согласно подпункта 1 пункта 8 Порядка, устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение. Далее, согласно подпункта 1 пункта 8 Порядка, формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если: - в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Сравнение характеристики композиции по независимому п. 1 формулы дополнительного патента и характеристики композиции лекарственного средства Б*** показывает следующее.
В п. 1 формулы дополнительного патента охарактеризовано мультидозовое ингаляторное устройство для сухого порошка, содержащее единственный активный агент
- аклидиний, и откалиброванное для обеспечения введения после его задействования отмеренной номинальной дозы аклидиния, эквивалентной 400 мкг (плюс/минус 10%) аклидиния бромида.
В РУ (раздел "Форма выпуска") и Инструкции (см. стр. 1 раздел "Ингалятор") охарактеризовано мультидозовое (на 30 или 60 доз) ингаляторное устройство, в которое помещено лекарственное средство Б*** в форме порошка, содержащее один активный ингредиент - аклидиния бромид. Одна доставленная доза аклидиния с помощью данного устройства составляет 322 мкг, что эквивалентно отмеренной номинальной дозе в 400 мкг аклидиния бромида.
Таким образом, независимый п. 1 представленной патентообладателем формулы дополнительного патента характеризует продукт, относящийся к лекарственному средству Б***, на применение которого получено разрешение, поскольку совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, указанного в разрешении.
Таким образом, несмотря на то, что ингалятор не относится к лекарственному средству, как таковому, отказать в продлении исключительного права в данном случае не представляется возможным, ввиду того, что и в РУ и в формуле изобретения дополнительного патента объект охарактеризован идентично. Использование лекарственной композиции невозможно без применения ингалятора. Дополнительный патент может быть выдан с формулой изобретений, представленной патентообладателем совместно с ходатайством (заявление) о продлении срока действия исключительного права на изобретение.
2.3.10. Уточнение радикалов в структурной формуле (формуле Маркуша), в соответствии с продуктом, на который получено Регистрационное удостоверение.
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Соединения формулы (I)
Рисунок (не приводится)
где R1 означает группу -NR3C(=O)OR4,
R2 означает атом водорода или NH2,
R3 означает атом водорода или алкильную группу с числом атомов углерода от одного до четырех,
R4 означает алкильную группу с числом атомов углерода от одного до шести, а также их соли, изомеры и гидраты.
2. Соединения по п. 1,
где R1 означает группу -NR3C(=O)OR4,
R2 означает атом водорода или NH2,
R3 означает алкильную группу с числом атомов углерода от одного до четырех,
R4 означает алкильную группу с числом атомов углерода от одного до четырех, а также их соли, изомеры и гидраты.
3. Соединения по п. 1,
где R1 означает группу -NR3C(=O)OR4,
R2 означает NH2,
R3 означает метильную или этильную группу,
R4 означает метильную, этильную или изопропильную группу, а также их соли, изомеры и гидраты.
4. Соединение по п. 1, представляющее собой метиловый эфир 4,6-диамино-2-[1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]-5-пиримидинил(метил)карбаминовой кислоты
Патентообладатель представил Регистрационное удостоверение N ЛП-000001, которое выдано на лекарственное средство А***, включающее в качестве активного начала химическое соединение, имеющее наименование метиловый эфир 4,6-диамино-2-[1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]-5-пиримидинил(метил)карбаминовой кислоты.
В рассматриваемом патенте RU*** вещество, указанное в Регистрационном удостоверении N ЛП-000001 получено в одном из примеров.
Пункты формулы изобретения RU*** имеют более широкие притязания в части значений радикалов, поэтому дополнительный патент может быть выдан только на конкретные радикалы, соответствующие активному веществу, на которое выдано Регистрационное удостоверение.
Таким образом, дополнительный патент может быть выдан со следующей совокупностью признаков:
Вариант 1
"Соединение формулы I
Рисунок (не приводится)
где
R1 означает группу -NR3C(=O)OR4,
R2 означает NH2,
R3 означает алкильную группу с одним атомом углерода,
R4 означает алкильную группу с одним атомом углерода".
Также данное соединение может быть представлено в форме варианта 2
Вариант 2
"Соединение формулы I
Рисунок (не приводится)
R1 означает группу -NR3C(=O)OR4,
R2 означает NH2,
R3 означает метильную группу,
R4 означает метильную группу".
Также данное соединение может быть представлено в форме варианта 3
Вариант 3
"Соединение формулы I, являющееся метиловым эфиром 4,6-диамино-2-[1-(2-фторбензил)-Ш-пиразоло[3,4-b]пиридин-3-ил]-5-пиримидинил(метил)карбаминовой кислоты.
2.3.11. Рассмотрение возможности включения в формулу дополнительного патента соединения и его соли.
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Производное оксазолидинона формулы I или его фармацевтически приемлемая соль
Рисунок (не приводится)
R1 и R1 независимо обозначают водород или фтор;
R2 обозначает -OR7, фтор, монофосфат или металлическую соль фосфата; и
R7 обозначает водород, С1-3алкил или ацилированную аминокислотную группу, где аминокислотой является аланин, глицин, пролин, изолейцин, лейцин, фенилаланин, Р-аланин или валин;
R3 обозначает водород, С1-4алкильную группу, которая является незамещенной или замещенной циано, -(CH2)m-OR7 (m равен 0, 1, 2, 3, 4) или кетогруппой.
Регистрационное удостоверение N ЛП-000001 выдано на лекарственный препарат С*** (тедизолид фосфат) активным ингредиентом которого является соединение, имеющее химическое название моно-[(R)-[3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-2-оксо-5-оксазолидинил]метил]фосфат.
Моно-[(R)-[3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-2-оксо-5-оксазолидинил]метил]фосфат в патенте RU*** раскрыт и получен в одном из приведенных примеров описания изобретения.
В случае, если в выданном Регистрационном удостоверении в качестве активного ингредиента указано вещество в виде его конкретной соли, то дополнительный патент может быть выдан с формулой изобретения, в которой должна быть указана данная соль.
Если в выданном Регистрационном удостоверении в качестве активного ингредиента указано вещество в виде основания, а в разделе Регистрационного удостоверения - "Состав лекарственного средства" указано, что активное вещество используется в виде его фармацевтически приемлемой соли, то дополнительный патент может быть выдан с формулой изобретения, в которой может быть указано само вещество, а также его фармацевтически приемлемая соль.
В данном примере в Регистрационном удостоверении N ЛП-000001 в качестве активного ингредиента указано вещество в виде его конкретной соли, поэтому дополнительный патент может быть выдан только на указанную конкретную соль.
Дополнительный патент может быть выдан на следующую совокупность признаков:
Производное оксазолидинона формулы I
Рисунок (не приводится)
где
R1 и R2 независимо обозначают водород или фтор;
R2 обозначает монофосфат;
R3 обозначает метил.
2.3.12. Рассмотрение возможности выдачи дополнительного патента на структурные производные (кристаллы, полиморфы и т.д.)
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Кристаллическая форма метансульфоната 4-(3-хлор-4-(циклопропиламинокарбонил)-аминофенокси)-7-метокси-6-хинолинкарбоксамида (форма A), имеющая дифракционные пики при углах отклонения при дифракции (29  0,2°) 9,65 и 18,37° в порошковой рентгенограмме.
0,2°) 9,65 и 18,37° в порошковой рентгенограмме.
2. Кристаллическая форма метансульфоната 4-(3-хлор-4-(циклопропиламинокарбонил)-аминофенокси)-7-метокси-6-хинолинкарбоксамида (форма A), имеющая пики при химических сдвигах примерно 162,4 м.д., примерно 128,0 м.д., примерно 102,3 м.д. и примерно 9,9 м.д. в спектре С-Ядерного Магнитного Резонанса в твердом состоянии.
3. Кристаллическая форма метансульфоната 4-(3-хлор-4-(циклопропиламинокарбонил)-аминофенокси)-7-метокси-6-хинолинкарбоксамида (форма A), имеющая полосы поглощения при волновых числах 1161 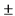 1 и 1044
1 и 1044  1 см-1 в инфракрасном спектре поглощения.
1 см-1 в инфракрасном спектре поглощения.
Регистрационное удостоверение N ЛП-000001 представлено на лекарственный препарат Л***, который имеет химическое название 4-(3-хлор-4-(циклопропиламинокарбонил)-аминофенокси)-7-метокси-6-хинолинкарбоксамид в виде его мезилатной (метансульфонатной) соли. В патенте RU*** данное соединение раскрыто в одном из примеров и на фигуре*** описания изобретения.
При этом, в Регистрационном удостоверении N ЛП-000001 нет указаний на то, что активный ингредиент препарата Л*** используется в виде кристаллической формы с определенными физико-химическими показателями. Инструкция к медицинскому применению препарата Л*** также не содержит таких сведений.
Для решения вопроса о продлении исключительного права у заявителя может быть запрошена дополнительные документы, например, декларация патентообладателя или производителя такого зарегистрированного препарата, в которой подтверждается, что вещество входит в препарат именно в кристаллической форме с указанными характеристиками этой формы. В случае если такая декларация имеется и характеристики препарата, на который выдано Регистрационное удостоверение N ЛП-000001 совпадают с характеристиками, указанными в одном из вариантов формулы изобретения по патенту RU***, исключительное право может быть продлено в отношении конкретной кристаллической формы с указанными характеристиками.
2.3.12. Рассмотрение возможности выдачи дополнительного патента в том случае, если рассматривается группа соединений, из которых только на одно имеется Регистрационное удостоверение; Идентичные дополнительные патенты
Патент RU(1)*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Моноклональное антитело или его антигенсвязывающая часть, содержащее вариабельную область тяжелой цепи, которая включает CDR1, CDR2 и CDR3 домены, и вариабельную область легкой цепи, которая включает CDR1, CDR2 и CDR3 домены, выбранные из:
(i) CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:15; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:22; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:29; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:36; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:43; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:50;
(ii) CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:16; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:23; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:30; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:37; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:44; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:51;
(iii) CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:18; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:25; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:32; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:39; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:46; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:53;
(iv) CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:17; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:24; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:31; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:38; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:45; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:52;
(vi) CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:20; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:27; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:34; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:41; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:48; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:55; и при этом моноклональное антитело или антитело или его антигенсвязывающая часть связывается с PD-1 человека с KD 1 · 10-7 M или менее, определенной с помощью анализа Biacore.
2. Моноклональное антитело или его антигенсвязывающая часть по п. 1, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью, выбранной из SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 и SEQ ID NO:13, и/или вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью, выбранной из SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 и SEQ ID NO:6.
3. Моноклональное антитело или его антигенсвязывающая часть по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 85% идентичности с последовательностью, выбранной из SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 и SEQ ID NO:13, и/или вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 85% идентичности с последовательностью, выбранной из SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 и SEQ ID NO: 6.
4. Моноклональное антитело или его антигенсвязывающая часть по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 90% идентичности с последовательностью, выбранной из SEQ ID NO: , SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO:11 и SEQ ID NO:13, и/или вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 90% идентичности по последовательности с последовательностью, выбранной из SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4 и SEQ ID NO:6.
5. Моноклональное антитело или его антигенсвязывающая часть по п. 1, отличающееся тем, что последовательности каркасных областей VH получены из последовательности VH 3-33 зародышевой линии человека, последовательности VH JH4b зародышевой линии человека, последовательности VH 7-27 зародышевой линии человека, последовательности VH 4-39 зародышевой линии человека или последовательности VH 39 зародышевой линии человека; или последовательности каркасных областей VL получены из последовательности VK L6 зародышевой линии человека, последовательности VK JK4 зародышевой линии человека, последовательности VK JK1 зародышевой линии человека или последовательности VK L15 зародышевой линии человека.
6. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое связывается с PD-1 человека с KD 0,5 · 10-8 М или менее, определенной с помощью анализа Biacore.
7. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое связывается с PD-1 человека с KD 1 · 10-9 М или менее, определенной с помощью анализа Biacore.
8. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое содержит:
(a) CDR1 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID NO:18;
(b) CDR2 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID NO:25;
(c) CDR3 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID NO:32;
(d) CDR1 легкой цепи, содержащий аминокислотную последовательность SEQ ID NO:
39;
(e) CDR2 легкой цепи, содержащий аминокислотную последовательность SEQ ID NO:46; и
(f) CDR3 легкой цепи, содержащий аминокислотную последовательность SEQ ID NO:53.
...
13. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, которое является гуманизированным антителом или антителом человека или его антигенсвязывающей частью.
14. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, которое ингибирует супрессию CD4+CD25-Т-клеток регуляторными Т-клетками.
15. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, которое усиливает антиген-специфичную ответную реакцию памяти на опухоль или патоген.
16. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, которое обеспечивает постоянный иммунитет к опухолевому рецидиву.
17. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, которое представляет собой антитело IgG1 или IgG4 или его часть.
18. Моноклональное антитело или его антигенсвязывающая часть по п. 15, которое представляет собой IgG4 и не обладает ни ADCC активностью, ни CDC активностью.
19. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 12, где его антигенсвязывающая часть выбрана из фрагмента Fab, F(ab')2, Fd, Fv или dAb, CDR или одноцепочечного Fv (scFv).
...
23. Композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество:
(a) моноклонального антитела или его антигенсвязывающей части по любому из пп. 1 - 12;
(b) иммуноконъюгата по п. 20; или
(c) биспецифического антитела или его антигенсвязывающей части по п. 22;
при этом указанная композиция предназначена для модуляции иммунной реакции.
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых и зависимых пунктах 1, 23 и зависимых пп. 2 - 8, 13 - 19 патента RU(1)***.
Ходатайство было рассмотрено с учетом действующего законодательства на момент его подачи, а именно: ст. 1363 Гражданского Кодекса РФ, Административного регламента предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение (утв. приказом Минэкономразвития России от 3 ноября 2015 года N 810) и удостоверяющего это право патента и Порядка выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение (утв. приказом Минэкономразвития России от 3 ноября 2015 года N 809).
В соответствии с п. 16 Административного регламента патентообладатель предоставил следующие документы:
1) заявление на 2 л.;
2) доводы патентообладателя на 9 л.;
3) копия Регистрационного удостоверения N ЛП-000001 от 01.01.2017 на 1 л.;
4) скорректированная формула изобретения для получения дополнительного патента на 3 л.;
5) копия МНН ВОЗ (WHO Drug Information, Vol. 26, No. 2, 2012, Proposed INN: List 107, "International N onproprietary N ames for Pharmaceutical Substances (INN)", стр. 157, 190, 191) на 2 л.;
6) копия инструкции по применению лекарственного препарата для медицинского применения О***, 20 л.
Регистрационного удостоверения N ЛП-000001 от 01.01.2017 выдано впервые со сроком действия 5 лет. Таким образом, оно соответствует требованиям п/п. 2) п. 8 Порядка.
Скорректированная формула изобретения, предоставленная патентообладателем для получения дополнительного патента и основанная на пп. 1 - 8, 13 - 19, 23 патента RU(1)***, имела следующий вид:
1. Моноклональное антитело или его антигенсвязывающая часть, содержащее вариабельную область тяжелой цепи, которая включает CDR1, CDR2 и CDR3 домены, и вариабельную область легкой цепи, которая включает CDR1, CDR2 и CDR3 домены:
CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:18; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:25; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:32; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:39; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:46; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:53; и при этом моноклональное антитело или антитело или его антигенсвязывающая часть связывается с PD-1 человека с KD 1 · 10-7 M или менее, определенной с помощью анализа Biacore.
2. Моноклональное антитело или его антигенсвязывающая часть по п. 1, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью SEQ ID NO:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью SEQ ID N0:4.
3. Моноклональное антитело или его антигенсвязывающая часть по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 85% идентичности с последовательностью, выбранной из SEQ ID NO:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 85% идентичности с последовательностью SEQ ID NO:4.
4. Моноклональное антитело или его антигенсвязывающая часть по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 90% идентичности с последовательностью SEQ ID N0:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 90% идентичности по последовательности с последовательностью SEQ ID N0:4.
5. Моноклональное антитело или его антигенсвязывающая часть по п. 1, отличающееся тем, что последовательности каркасных областей VH получены из последовательности VH 3-33 зародышевой линии человека; или последовательности каркасных областей VL получены из последовательности VKL6 зародышевой линии человека.
6. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое связывается с PD-1 человека с KD 0,5 · 10-8 M или менее, определенной с помощью анализа Biacore.
7. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое связывается с PD-1 человека с KD 1 · 10-9 M или менее, определенной с помощью анализа Biacore.
8. Моноклональное антитело или его антигенсвязывающая часть по п. 1, которое содержит:
CDR1 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N 0:18;
CDR2 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N 0:25;
CDR3 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N 0:32;
CDR1 легкой цепи, содержащий аминокислотную последовательность SEQ ID N 0:39;
CDR2 легкой цепи, содержащий аминокислотную последовательность SEQ ID N 0:46; и
CDR3 легкой цепи, содержащий аминокислотную последовательность SEQ ID N 0:53.
9. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, которое является гуманизированным антителом или антителом человека или его антигенсвязывающей частью.
10. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, которое ингибирует супрессию CD4+CD25-Т-клеток регуляторными Т-клетками.
11. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, которое усиливает антиген-специфичную ответную реакцию памяти на опухоль или патоген.
12. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, которое обеспечивает постоянный иммунитет к опухолевому рецидиву.
13. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, которое представляет собой антитело IgG1 или IgG4 или его часть.
14. Моноклональное антитело или его антигенсвязывающая часть по п. 13, которое представляет собой IgG4 и не обладает ни ADCC активностью, ни CDC активностью.
15. Моноклональное антитело или его антигенсвязывающая часть по любому из пп. 1 - 8, где его антигенсвязывающая часть выбрана из фрагмента Fab, F(ab')2, Fv или dAb, CDR или одноцепочечного Fv (scFv).
16. Композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество:
(a) моноклонального антитела или его антигенсвязывающей части по любому из пп. 1 - 8;
при этом указанная композиция предназначена для модуляции иммунной реакции.
Согласно п. 2 ст. 1363 Кодекса, а также п. 16 и п. 21 Административного регламента продление срока действия исключительного права на изобретение осуществляется в отношении изобретения, относящегося к лекарственному средству, пестициду или агрохимикату.
Согласно п/п. 1) п. 8 Порядка формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если:
- в формуле изобретения продукт охарактеризован в виде соединения или группы соединений, описываемых общей структурной формулой, и из описания изобретения следует возможность его (ее) использования в качестве активного ингредиента лекарственного средства, пестицида или агрохимиката, при этом совокупность признаков, определяющих объем правовой охраны продукта, характеризующих соединение или группу соединений, описываемых общей структурной формулой, идентична активному ингредиенту лекарственного средства, пестицида или агрохимиката, указанного в полученном разрешении на применение этого продукта, а описание изобретения содержит информацию о том, что соединение или группа соединений, описываемых общей структурной формулой, обладает такой активностью, которая позволяет его (ее) использовать в указанном в разрешении лекарственном средстве, пестициде или агрохимикате;
- в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции).
Характеристика лекарственного препарата О***, указанная в представленном РУ N ЛП-000001 от 01.01.2017 включает следующие признаки: Ниволумаб 47,0 мг или 107.0 мг (активное вещество) и вспомогательные вещества.
Ниволумаб, согласно МНН ВОЗ, имеет следующие характеристики:
inmunoglobulina G4-kappa, anti-[PDCD1 de Homo sapiens (proteina 1 de muerte celular programada, PD-1, PD1, CD279)], anticuerpo monoclonal de Homo sapiens;
cadena pesada gammal (1-440) [Homo sapiens VH (IGHV3-33*01 (91.80%)-(IGHD)-IGHJ4*01) [8.8.6] (1-113)-IGHG4*01 bisagra S10 > P (221) (114-440)], (127-214')-disulfuro con la cadena ligera kappa (1-214') [Homo sapiens V-KAPPA (IGKV3-11*01 (98.90%) - IGKJ1*01) [6.3.9J (1'-107')-IGKC*01 (108-214*)];
dimero (219-219":222-222")-bisdisulfuro inmunomodulador
946414-94-4
|
Heavy chain/Chaine lourde/Cadena pcsada
|
|||||
|
QVOLVESGGG
|
WQPGRSLRL
|
DCKASGITFS
|
N SGMHWVRQA
|
PGKGLEWVAV
|
50
|
|
IWYDGSKRYY
|
ADSVKGRFTI
|
SRDN SKN TLF
|
LQMN SLRAED
|
TAVYYCATN D
|
100
|
|
DYWGQGTLVT
|
VSSASTKGPS
|
VFPLAPCSRS
|
TSESTAALGC
|
LVKDYFPEPV
|
150
|
|
TVSWN SGALT
|
SGVHTFPAVL
|
QSSGLYSLSS
|
WTVPSSSLG
|
TKTYTCN VDH
|
200
|
|
KPSN TKVDKR
|
VESKYGPPCP
|
PCPAPEFLGG
|
PSVFLFPPKP
|
KDTLMISRTP
|
250
|
|
EVTCVWDV5
|
QEDFEVQFN W
|
YVDGVEVHN A
|
KTKFREEQFN
|
5TYRWSVLT
|
300
|
|
VLHQDWLN GK
|
EYKCKVSN KG
|
LPSSIEKTIS
|
KAKGQPREPQ
|
VYTLPPSQEE
|
350
|
|
MTKN QVSLTC
|
LVKGFYPSDI
|
AVEWESN GQP
|
EN N YKTTPPV
|
LDSDGSFFLY
|
400
|
|
SRLTVDKSRW
|
QEGN VFSCSV
|
MHEALHN HYT
|
QKSLSLSLGK
|
440
|
|
|
Light chain/Chaine I6gere/Cadena ligera
|
|||||
|
EIVLTQSPAT
|
LSLSPGERAT
|
LSCRASQSVS
|
SYLAWYQQKP
|
GQAPRLL1YD
|
50
|
|
ASN RATGIPA
|
RFSGSGSGTD
|
FTLT1SSLEP
|
EDFAVYYCQQ
|
SSN WPRTFGQ
|
100
|
|
GTKVEIKRTV
|
AAPSVFIFPP
|
SDEQLKSGTA
|
SWCLLN N FY
|
PREAKVQWKV
|
150
|
|
DN AI.QSGN SQ
|
ESVTEQDSKD
|
STYSLSSTLT
|
LSKADYEKHK
|
VYACEVTHQG
|
200
|
|
LSSPVTKSFN
|
RGEC
|
214
|
|||
|
Disulfide bridges location/Position des ponts disulfure/Posiciones de los puentes disulfuro
|
||||
|
lntia-H
|
22 - 96
|
140 - 196
|
254 - 314
|
360 - 418
|
|
2
|
2" - 96"
|
140" - 196"
|
254" - 314"
|
360" - 418"
|
|
lntra-L
|
23' - 88'
|
134' - 194'
|
||
|
2
|
23 "' - 88"'
|
134"' - 194"'
|
||
|
Inter-H-L
|
127 - 214'
|
127" - 214"
|
||
|
Inter-H-H
|
219 - 219"
|
222 - 222"
|
||
|
N-gfycosylation sites/Sites de N-glycosylation/Posiciores de N-glicosilacion
|
||||
|
HCH2 84.4
290, 290"
|
||||
Согласно Инструкции, лекарственный препарат О*** применяется для лечения меланомы, немелкоклеточного рака легких или почечно-клеточного рака.
Сравнение характеристик продукта по п. 1, и характеристики активного ингредиента лекарственного средства О*** (Ниволумаб) показало следующее.
Последовательности CDR активного ингредиента лекарственного препарата О*** (Ниволумаб) и последовательности CDR антитела и антигенсвязывающей части антитела по п. 1 идентичны. Однако Ниволумаб представляет собой полноразмерное человеческое антитело, тогда как п. 1 формулы изобретения, предложенной патентообладателем для получения дополнительного патента, охватывает антигенсвязывающие части и антитела, аминокислотные последовательности которых, за исключением CDR-участков, могут иметь происхождение любых биологических видов. Возможность использования в качестве активного ингредиента каких-либо иных антител, а также их фрагментов, за исключением полноразмерного человеческого антитела, не указывалась в Регистрационном удостоверении N ЛП-000001 от 01.01.2017 и приложенных к нему документах. Иными словами, в п. 1 заявлена группа соединений, из которых только на одно получено разрешение.
Согласно п/п. 1) п. 8 Порядка при проверки того, характеризует ли формула изобретения продукт, который относится к лекарственному средству и на применение которого получено разрешение, необходимо, чтобы описание изобретения содержало информацию о том, что соединение или группа соединений, описываемых общей структурной формулой, обладали такой активностью, которая позволяет использовать их в лекарственном средстве, указанном в разрешении.
В данном случае в описании патента на изобретение не приведены сведения о том, что антитела, имеющие аминокислотные последовательности, которые принадлежат видам, отличным от человека, а также антигенсвязывающие части антитела обладают активностью, которая позволяет их использовать в качестве активного ингредиента лекарственного препарата, на которое получено разрешение. Возможность применения в качестве активного ингредиента лекарственного средства показана в материалах заявки только для антитела, которое обладает константной областью антитела человека, в связи с чем проявляет эффекторные свойства при использовании для лечения человека. Учитывая, что для специалиста в данной области известно, что антигенсвязывающие части антитела не имеют константных доменами и, как следствие, не способны проявлять эффекторные функции, они не обладают активностью, позволяющей использовать их в качестве активного ингредиента лекарственного средства, на которое получено разрешение. Антитела, имеющие константные области иных видов, отличных от человека, как правило, также не способны проявлять эффекторных функций при использовании их для лечения человека, поскольку имеющие у человека рецепторы эволюционно приспособлены связывать только константные области человеческих антител.
Таким образом, установлено, что п. 1 не соответствует требованиям п/п. 1) п. 8 Порядка.
Сравнение характеристик продукта по п. 16 формулы изобретения, предоставленной патентообладателем для получения дополнительного патента, и характеристики лекарственного средства О***, на которое получено Регистрационном удостоверении N ЛП-000001 от 01.01.2017, показало следующее.
Согласно п/п. 1) п. 8 Порядка совокупность признаков, определяющих объем правовой охраны продукта, характеризующего композицию, указанную в формуле изобретения, должна быть идентична характеристике композиции лекарственного средства, указанной в разрешении, в том числе назначением.
Согласно п. 16 формулы изобретения, предложенной патентообладетелем для получения дополнительного патента, продукт представляет собой композицию, содержащую фармацевтически приемлемый носитель и терапевтически эффективное количество моноклонального антитела или его антигенсвязывающей части по любому из пп. 1 - 8; при этом указанная композиция предназначена для модуляции иммунной реакции.
Согласно предоставленным патентообладателем документам (стр. 3 инструкции), лекарственный препарат О*** предназначен только для лечения меланомы, немелкоклеточного рака легких и почечно-клеточного рака. Назначение для лечения других заболеваний не указано ни в Регистрационном удостоверении N ЛП-000001 от 01.01.2017, ни в приложенных к нему документах. Напротив, согласно стр. 13 - 18 Инструкции, в случаях назначения при других заболеваниях лекарственный препарат О*** может вызывать тяжелые побочные эффекты.
Таким образом, совокупность признаков, определяющих объем правовой охраны продукта по п. 16 скорректированной формулы изобретения для получения дополнительного патента, не идентична характеристики композиции лекарственного средства О*** (п/п. 1) п. 8 Порядка).
Заявителю было предложено надлежащим образом скорректировать формулу изобретения.
В данном случае дополнительный патент может быть выдан на следующую совокупность признаков:
1. Человеческое моноклональное антитело -и-л-и- -е-г-о а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь-, содержащее вариабельную область тяжелой цепи, которая включает CDR1, CDR2 и CDR3 домены, и вариабельную область легкой цепи, которая включает CDR1, CDR2 и CDR3 домены:
CDR1 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:18; CDR2 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:25; CDR3 тяжелой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:32; CDR1 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:39; CDR2 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:46; и CDR3 легкой цепи, содержащего аминокислотную последовательность, приведенную в SEQ ID NO:53; и
при этом моноклональное антитело -и-л-и- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- связывается с PD-1 человека с KD 1 · 10-7 M или менее, определенной с помощью анализа Biacore.
2. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 1, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью SEQ ID NO:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 80% идентичности с последовательностью SEQ ID N0:4.
3. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 85% идентичности с последовательностью, выбранной из SEQ ID NO:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 85% идентичности с последовательностью SEQ ID NO:4.
4. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 2, отличающееся тем, что вариабельная область легкой цепи содержит аминокислотную последовательность, имеющую по меньшей мере на 90% идентичности с последовательностью SEQ ID N0:11 и вариабельная область тяжелой цепи содержит аминокислотную последовательность, имеющую по меньшей мере 90% идентичности по последовательности с последовательностью SEQ ID N0:4.
5. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 1, отличающееся тем, что последовательности каркасных областей VH получены из последовательности VH 3-33 зародышевой линии человека; или последовательности каркасных областей VL получены из последовательности VK L6 зародышевой линии человека.
6. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 1, которое связывается с PD-1 человека с KD 0,5 · 10-8 M или менее, определенной с помощью анализа Biacore.
7. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-яз-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по п. 1, которое связывается с PD-1 человека с KD 1 · 10-9 M или менее, определенной с помощью анализа Biacore.
8. Человеческое моноклональное антитело -и-л-и- -е-го- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-ас-т-ь- по п. 1, которое содержит:
CDR1 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N0:18;
CDR2 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N0:25;
CDR3 тяжелой цепи, содержащий аминокислотную последовательность SEQ ID N0:32;
CDR1 легкой цепи, содержащий аминокислотную последовательность SEQ ID N0:39;
CDR2 легкой цепи, содержащий аминокислотную последовательность SEQ ID N0:46; и
CDR3 легкой цепи, содержащий аминокислотную последовательность SEQ ID N0:53.
-9-.- -М-о-н-о-к-ло-н-а-л-ь-н-о-е- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- -п-о- -л-ю-б-о-м-у- -и-з- -п-п-.- -1- --- -8-,- -к-о-т-о-р-о-е- -я-в-л-я--е-т-с-я- -г-у-м-а-н-и-з-и-р-о-в-а-н-н-ы-м- -а-н-т-и-т-е-л-о-м- -и-л-и- -а-н-т-и-т-е-л-о-м- -ч-е-л-о-в-е-к-а- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-е-й- -ч-а-с-т-ь-ю-.
10. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е--н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по любому из пп. 1 - 8, которое ингибирует супрессию CD4+CD25-Т-клеток регуляторными Т-клетками.
11. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по любому из пп. 1 - 8, которое усиливает антиген-специфичную ответную реакцию памяти на опухоль или патоген.
12. Человеческое моноклональное антитело -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- по любому из пп. 1 - 8, которое обеспечивает постоянный иммунитет к опухолевому рецидиву.
-1-3-.- -М-о-н-о-к-л-о-н-а-л-ь-н-о-е- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- -п-о- -л-ю-б-о-м-у- -и-з- -п-п-.- -1- --- -8-,- -к-о-т-о-р-о-е- -п-р-е-д-с-т-а-в-л-я-е-т- -с-о-б-о-й- -а-н-т-и-т-е-л-о- -I-g-G-1- -и-л-и- -I-g-G-4- -и-л-и- -е-г-о- -ч-а-с-т-ь-.
-1-4-.- -М-о-н-о-к-л-о-н-а-л-ь-н-о-е- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- -п-о- -п-.- -1-3-,- -к-о-т-о-р-о-е- -п-р-е-д-с-т-а-в-л-я-е-т- -с-о-б-о-й- -I-g-G-4- -и- -н-е- -о-б-л-а-д-а-е-т- -н-и- -A-D-C-C- -а-к-т-и-в-н-о-с-т-ь-ю-,- -н-и- -C-D-C- -а-к-т-и-в-н-о-с-т-ь-ю-.
-1-5-.- М-М-о-н-о-к-л-о-н-а-л-ь-н-о-е- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- -п-о- -л-ю-б-о-м-у- -и-з- -п-п-. -1- --- -8-,- -г-д-е- -е-г-о- -а-н-т-и-г-е-н-с-в-я-з-ы-в-а-ю-щ-а-я- -ч-а-с-т-ь- -в-ы-б-р-а-н-а- -и-з- -ф-р-а-г-м-е-н-т-а- -F-a-b-,- -F-(-a-b-')-2-,- -F-v- -и-л-и- -d-A-b-,- -C-D-R- -и-л-и- -о-д-н-о-ц-е-п-о-ч-еч-н-о-г-о- -F-v- -(-s-c-F-v-)-.
16. Композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество:
(a) моноклонального антитела или его антигенсвязывающей части по любому из пп. 1 - 8:
при этом указанная композиция предназначена -д-л-я- -м-о-д-у-л-я-ц-и-и- -и-м-м-ун-н-о-й- -р-е-а-к-ц-и-и- для лечения меланомы, немелкоклеточного рака легких или почечно-клеточного рака.
Одновременно с заявлением о продлении срока действия исключительного права патента RU(1)***патентообладатель подал заявление о продлении срока действия исключительного права на изобретения, охарактеризованные в независимых пунктах 1 - 3 формулы изобретения патента RU(2)***, и предоставил вместе с ним то же самое Регистрационного удостоверения N ЛП-000001 от 01.01.2017.
Патент RU(2)*** был зарегистрирован с формулой содержащей следующую совокупность признаков:
1. Выделенное моноклональное антитело или его антигенсвязывающая часть, где антитело связывается с PD-1 и проявляет все перечисленные ниже свойства:
(a) связывается с PD-1 человека с KD 1 · 10<-8> М или менее;
(b) по существу, не связывается с CD28, CTLA-4 и ICOS человека;
(c) увеличивает пролиферацию Т-клеток в анализе реакции лимфоцитов в смешанной культуре (MLR);
(d) увеличивает продукцию интерферона-гамма в анализе MLR и
(e) увеличивает секрецию интерлейкина-2 (IL-2) в анализе MLR.
2. Выделенное моноклональное антитело или его антигенсвязывающая часть, где антитело выбрано из группы, состоящей из:
a) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:15;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:22;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:29;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:36;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:43, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:50;
b) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:16;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:23;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:30;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:37;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:44, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:51;
c) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:17;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:24;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:31;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:38;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:45, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:52;
d) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:18;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:25;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:32;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:39;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:46, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:53;
e) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:19;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:26;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:33;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:40;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:47, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:54;
f) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:20;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:27;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:34;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:41;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:48, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:55; и
g) антитела, содержащего
CDR1 вариабельной области тяжелой цепи, содержащей SEQ ID NO:21;
CDR2 вариабельной области тяжелой цепи, содержащей SEQ ID NO:28;
CDR3 вариабельной области тяжелой цепи, содержащей SEQ ID NO:35;
CDR1 вариабельной области легкой цепи, содержащей SEQ ID NO:42;
CDR2 вариабельной области легкой цепи, содержащей SEQ ID NO:49, и
CDR3 вариабельной области легкой цепи, содержащей SEQ ID NO:56;
где антитело специфически связывает PD-1.
3. Выделенное моноклональное антитело или его антигенсвязывающая часть, где антитело выбрано из группы, состоящей из:
a) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID N 0:1, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:8;
b) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:2, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:9;
c) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:3, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:10;
d) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:4, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:11;
e) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:5, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:12;
f) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:6, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:13; и
g) антитела, содержащего вариабельную область тяжелой цепи, содержащую аминокислотную последовательность SEQ ID NO:7, и вариабельную область легкой цепи, содержащую аминокислотную последовательность SEQ ID NO:14, где антитело специфически связывает PD-1.
...
По аналогии с предыдущим патентом RU(1)*** патентообладателю было сообщено на несоответствие пп. 1 - 3 требованиям п/п. 1) п. 8 Порядка, поскольку пункты 1 - 3 формулы изобретения патента RU(1)***, также как и п. 1 формулы патента RU(2)*** охватывает моноклональное антитело, имеющее аминокислотные последовательности любых биологических видов, а также антигенсвязывающие части антитела, не содержащие константных доменов и возможность использования в лекарственном средстве, указанном в разрешении, которых не подтверждена описанием изобретения.
При этом формула изобретения, с которой может быть выдан дополнительный патент RU(2)*** в данном случае, оказывается идентичной формуле, с которой может быть выдан дополнительный патент к патенту RU(1)*** в части независимого п. 1.
Исходя из требований статьи 1383 Гражданского Кодекса в случае установления идентичности независимых пунктов, содержащихся в разных патентах, патентообладателю предстоит выбрать, в отношении какого из двух указанных патентов RU(1)*** или RU(2)*** продолжить делопроизводство и принять решение о продлении исключительного права путем выдачи дополнительного патента.
2.3.13. Рассмотрение возможности выдачи дополнительного патента, в котором осуществляется уточнение назначения лекарственного средства; рассмотрение ответа патентообладателя.
Патент RU*** зарегистрирован с формулой содержащей следующую совокупность признаков:
1. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина, где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1, CDR2 и CDR3, где CDR1 имеет аминокислотную последовательность SEQ ID NO:1, CDR2 имеет аминокислотную последовательность SEQ ID NO:2 и CDR3 имеет аминокислотную последовательность SEQ ID NO:3, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6, причем CDR последовательность соответствует обозначению Kabat.
2. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина, где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1-x, CDR2-x и CDR3-x, где CDR1-x имеет аминокислотную последовательность SEQ ID NO:11, CDR2-x имеет аминокислотную последовательность SEQ ID NO:12 и CDR3-x имеет аминокислотную последовательность SEQ ID No:13, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6, причем CDR последовательность соответствует обозначению Chotia.
3. IL-17-связывающее антитело или его активный фрагмент по п. 1 или п. 2, представляющее собой человеческое антитело.
4. IL-17-связывающее антитело или его активный фрагмент, которое содержит по меньшей мере один антигенсвязывающий центр, включающий первый домен, имеющий аминокислотную последовательность, представленную в SEQ ID NO:8, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 127, или представленную в SEQ ID NO:10, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 109.
9. Применение IL-17-связывающего антитела или его активного фрагмента по любому из п. п. 1 - 4 для получения лекарственного средства для лечения опосредуемого IL-17 заболевания или нарушения.
10. Применение IL-17-связывающего антитела или его активного фрагмента по любому из п. п. 1 - 4 для лечения псориаза, увеита, остеоартрита, ревматоидного артрита, остеопороза и других воспалительных поражений кожи ревматического или подагрического происхождения.
11. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из п. п. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения опосредуемого IL-17 заболевания или нарушения.
12. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из п. п. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения псориаза, увеита, остеоартрита, ревматоидного артрита, остеопороза и других воспалительных поражений кожи ревматического или подагрического происхождения.
...
От патентообладателя поступило ходатайство о продлении срока действия патента RU*** на изобретение, охарактеризованное в независимых пунктах 1, 2, 4, 9, 10, 11 и 12 и зависимом п. 3 формулы изобретения патента и выдачи дополнительного патента.
Ходатайство было рассмотрено с учетом действующего законодательства на момент его подачи, а именно: ст. 1363 Гражданского Кодекса РФ, Административного регламента предоставления Федеральной службой по интеллектуальной собственности государственной услуги по продлению срока действия исключительного права на изобретение и удостоверяющего это право патента (утв. приказом Минэкономразвития России от 3 ноября 2015 года N 810) и Порядка выдачи и действия дополнительного патента на изобретение, продление срока действия патента на изобретение (утв. приказом Минэкономразвития России от 3 ноября 2015 года N 809).
В соответствии с п. 16 Административного регламента патентообладатель предоставил следующие документы:
1) заявление с приложением, включающим доводы заявителя, на 5 л.;
2) копия Регистрационного удостоверения N ЛП-000001 от 01.01.2017 на 2 л.;
3) скорректированная формула изобретения для дополнительного патента на 2 л.;
4) приложение 1-WHO Drug Information, Vol. 24, No. 3, 2010, Recommended INN: List 64 "International N onproprietary N ames for Pharmaceutical Substances (INN)", стр. 278279, с переводом на 3 л.;
5) приложение 2 - Инструкция по применению лекарственного препарата К*** для медицинского применения, 13 л.
Регистрационного удостоверения N ЛП-000001 от 01.01.2017 выдано впервые со сроком действия 5 лет. Таким образом, оно соответствует требованиям п/п. 2) п. 8 Порядка.
Скорректированная формула изобретения, предоставленная патентообладателем для получения дополнительного патента и основанная на пп. 1 - 4, 9 - 12 патента N RU***, имела следующий вид:
1. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1, CDR2 и CDR3, где CDR1 имеет аминокислотную последовательность SEQ ID N0:1, CDR2 имеет аминокислотную последовательность SEQ ID N0:2 и CDR3 имеет аминокислотную последовательность SEQ ID N0:3, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID N0:4, CDR2' имеет аминокислотную последовательность SEQ ID N0:5 и CDR3' имеет аминокислотную последовательность SEQ ID N0:6, причем CDR последовательность соответствует обозначению Kabat.
2. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDRl-x, CDR2-X и CDR3-X, где CDRl-x имеет аминокислотную последовательность SEQ ID N0:11, CDR2-X имеет аминокислотную последовательность SEQ ID N0:12 и CDR3-X имеет аминокислотную последовательность SEQ ID N0:13, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID N0:4, CDR2' имеет аминокислотную последовательность SEQ ID N0:5 и CDR3' имеет аминокислотную последовательность SEQ ID N0:6, причем CDR последовательность соответствует обозначению Chotia.
3. IL-17-связывающее антитело или его активный фрагмент по п. 1 или 2, представляющее собой человеческое антитело.
4. IL-17-связывающее антитело или его активный фрагмент, которое содержит по меньшей мере один антигенсвязывающий центр, включающий первый домен, имеющий аминокислотную последовательность, представленную в SEQ ID N0:8, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 127, или представленную в SEQ ID N0:10, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 109.
5. Применение IL-17-связывающего антитела или его активного фрагмента по любому из пп. 1 - 4 для получения лекарственного средства для лечения опосредуемого IL-17 заболевания или нарушения.
6. Применение IL-17-связывающего антитела или его активного фрагмента по любому из пп. 1 - 4 для лечения псориаза, увеита, остеоартрита, ревматоидного артрита, остеопороза и других воспалительных поражений кожи ревматического или подагрического происхождения.
7. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из пп. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения опосредуемого IL-17 заболевания или нарушения.
8. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из пп. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения псориаза, увеита, остеоартрита, ревматоидного артрита, остеопороза и других воспалительных поражений кожи ревматического или подагрического происхождения.
Характеристика лекарственного препарата К***, указанная в представленном Регистрационного удостоверения N ЛП-000001 от 01.01.2017 включает следующие признаки:
Действующие и вспомогательные вещества: Секукинумаб 150.00 мг, вспомогательные вещества.
Сравнение характеристик антитела, запатентованного в независимых пп. 1, 2, 4 и зависимом п. 3 формулы изобретения, предложенной патентообладателем для получения дополнительного патента, и активного ингредиента лекарственного средства К*** в соответствии с п/п. 1) п. 8 Порядка показало, что вышеуказанные пункты формулы изобретения идентичны характеристике активного ингредиента лекарственного средства, указанного в Регистрационного удостоверения N ЛП-000001 от 01.01.2017.
В Регистрационного удостоверения N ЛП-000001 от 01.01.2017 указано, что действующим веществом лекарственного средства К*** является Секукинумаб.
Согласно приложению к заявлению (выдержка из журнала "WHO Drug Information" Всемирной Организации Здравоохранения), CDR1 тяжелой цепи Секукинумаба имеет аминокислотную последовательность SEQ ID NO:1, CDR2 тяжелой цепи имеет аминокислотную последовательность SEQ ID NO:2 и CDR3 тяжелой цепи имеет аминокислотную последовательность SEQ ID NO:3, а CDR1' легкой цепи имеет аминокислотную последовательность SEQ ID NO:4, CDR2' легкой цепи имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' легкой цепи имеет аминокислотную последовательность SEQ ID NO:6. При этом описание изобретение содержит информацию о том, что вся группа соединений, описываемых общими признаками, указанными в пп. 1 - 4, обладают активностью, которая позволяет их использовать в качестве активного ингредиента лекарственного средства, указанного в Регистрационного удостоверения N ЛП-000001 от 01.01.2017.
Сравнение характеристик применения по пп. 5, 6, фармацевтический композиций по пп. 7, 8 с характеристиками лекарственного средства К*** показало следующее. Характеристика объектов по пп. 5, 7 включает указание назначения "для лечения опосредуемого IL-17 заболевания или нарушения"; характеристика объектов по пп. 6, 8 включает указание назначения "для лечения псориаза, увеита, остеоартрита, ревматоидного артрита, остеопороза и других воспалительных поражений кожи ревматического или подагрического происхождения". Согласно Регистрационного удостоверения N ЛП-000001 от 01.01.2017 и приложенным к нему документам, разрешение получено на применение лекарственного средства для лечения псориаза, активного псориатрического артрита и активного анкилозирующего спондилита. В этой связи сделан вывод о том, что пп. 5 - 8 не соответствуют требованию п/п. 1) п. 8 Порядка.
Патентообладателю был направлен запрос с просьбой предоставить надлежащим образом скорректированную формулу изобретения.
В ответ на запрос патентообладатель предложил следующую редакцию формулы:
1. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1, CDR2 и CDR3, где CDR1 имеет аминокислотную последовательность SEQ ID NO:1, CDR2 имеет аминокислотную последовательность SEQ ID NO:2 и CDR3 имеет аминокислотную последовательность SEQ ID NO:3, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6,
причем CDR последовательность соответствует обозначению Kabat.
2. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1-x, CDR2-x и CDR3-x, где CDR1-x имеет аминокислотную последовательность SEQ ID NO:11, CDR-x имеет аминокислотную последовательность SEQ ID NO:12 и CDR3-x имеет аминокислотную последовательность SEQ ID NO:13, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6, причем CDR последовательность соответствует обозначению Chotia.
3. IL-17-связывающее антитело или его активный фрагмент по п. 1 или 2, представляющее собой человеческое антитело.
4. IL-17-связывающее антитело или его активный фрагмент, которое содержит по меньшей мере один антигенсвязывающий центр, включающий первый домен, имеющий аминокислотную последовательность, представленную в SEQ ID N0:8, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 127, или представленную в SEQ ID N0:10, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 109.
5. Применение IL-17-связывающего антитела или его активного фрагмента по любому из пп. 1 - 4 для получения лекарственного средства для лечения опосредуемого IL-17 заболевания или нарушения.
6. Применение IL-17-связывающего антитела или его активного фрагмента по любому из пп. 1 - 4 для лечения псориаза, -у-в-е-и-т-а-,- -о-с-т-е-о-а-р-т-р-и-т-а-,- -р-е-в-м-а-т-о-и-д-н-о-г-о- -а-р-т-р-и-т-а-,- -о-с-т-е-о-п-о-р-о-з-а- -и- -д-р-у-г-и-х- -в-о-с-п-а-л-и-т-е-л-ь-н-ы-х- -п-о-р-а-ж-е-н-и-й- -к-о-ж-и- -р-е-в-м-а-т-и-ч-е-с-к-о-г-о- -и-л-и- -п-о-д-а-г-р-и-ч-е-с-к-о-г-о- -п-р-о-и-с-х-о-ж-д-е-н-и-я- псориатического артрита или анкилозирующего спондилита.
7. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из пп. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения опосредуемого IL-17 заболевания или нарушения.
8. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из пп. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения псориаза, -у-в-е-и-т-а-,- -о-с-т-е-о-а-р-т-р-и-т-а-,- -р-е-в-м-а-т-о-и-д-н-о-г-о- -а-р-т-р-и-т-а-,- -о-с-т-е-о-п-о-р-о-з-а- -и- -д-р-у-г-и-х- -в-о-с-п-а-л-и-т-е-л-ь-н-ы-х- -п-о-р-а-ж-е-н-и-й- -к-о-ж-и- -р-е-в-м-а-т-и-ч-е-с-к-о-г-о- -и-л-и- -п-о-д-а-г-р-и-ч-е-с-к-о-г-о- -п-р-о-и-с-х-о-ж-д-е-н-и-я- псориатического артрита или анкилозирующего спондилита.
В ходе проверки представленной патентообладателем измененной формулы изобретения было установлено следующее.
Согласно п/п. 1) п. 8 Порядка совокупность признаков, определяющих объем правовой охраны продукта, характеризующего композицию, указанную в формуле изобретения, должна быть идентична характеристике композиции лекарственного средства, указанной в разрешении, в том числе назначением.
Пункты 6, 8 формулы изобретения, предложенной патентообладателем в ответе, соответствуют указанным требованиям.
Однако характеристика объектов по п. 5 и п. 7 скорректированной формулы изобретения по-прежнему включает назначение для лечения любого опосредуемого IL-17 заболевания или нарушения. При этом в своем ответе патентообладатель указал, что "согласно Инструкции по применению, в частности, указанному там механизму действия Секукинумаба, указанное антитело ингибирует взаимодействие IL-17 с рецептором IL-17, чем понижается вклад IL-17 в опосредованные IL-17 аутоиммунные и воспалительные заболевания, и, таким образом, осуществляется их лечение". По мнению заявителя, данная активность позволяет использовать секукинумаб для лечения любого опосредуемого IL-17 заболевания или нарушения.
Между тем в ходе анализа Регистрационного удостоверения N ЛП-000001 от 01.01.2017 и приложенных к нему документов установлено, что разрешение получено на лекарственное средство только для лечения псориаза, активного псориатрического артрита и активного анкилозирующего спондилита. Показания для лечения других заболеваний или нарушений посредством лекарственного средства К*** не указаны ни в Регистрационного удостоверения N ЛП-000001 от 01.01.2017, ни в приложенной к нему Инструкции. Более того, в Инструкции по применению лекарственного препарата К*** для медицинского применения отмечено, что "в клинических исследованиях наблюдалось усугубление течения болезни Крона, в некоторых случаях тяжелое". Болезнь Крона, как известно, также является опосредованным IL-17 заболеванием.
Заявителю был направлен повторный запрос с указанием нарушения пп. 5, 7 требованию п/п. 1) п. 8 Порядка и с просьбой надлежащим образом скорректировать формулу изобретения. Между тем, учитывая, что в случае надлежащей корректировки пп. 5, 7 будут идентичны пп. 6, 8, патентообладателю целесообразно исключить пп. 5, 7 из формулы.
Учитывая вышеизложенное, дополнительный патент может быть выдан на следующую совокупность признаков:
1. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1, CDR2 и CDR3, где CDR1 имеет аминокислотную последовательность SEQ ID NO:1, CDR2 имеет аминокислотную последовательность SEQ ID NO:2 и CDR3 имеет аминокислотную последовательность SEQ ID NO:3, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID NO:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6, причем CDR последовательность соответствует обозначению Kabat.
2. IL-17-связывающее антитело или его активный фрагмент, содержащий вариабельные области как тяжелой цепи (VH), так и легкой цепи (VL) иммуноглобулина,
где IL-17-связывающее антитело или его активный фрагмент содержит по меньшей мере один антигенсвязывающий центр, включающий:
а) вариабельную область тяжелой цепи (VH) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDRl-x, CDR2-x и CDR3-x, где CDR1-x имеет аминокислотную последовательность SEQ ID No:11, CDR2-x имеет аминокислотную последовательность SEQ ID NO:12 и CDR3-x имеет аминокислотную последовательность SEQ ID NO:13, и
б) вариабельную область легкой цепи (VL) иммуноглобулина, которая содержит последовательно расположенные гипервариабельные участки CDR1', CDR2' и CDR3', где CDR1' имеет аминокислотную последовательность SEQ ID NO:4, CDR2' имеет аминокислотную последовательность SEQ ID No:5 и CDR3' имеет аминокислотную последовательность SEQ ID NO:6, причем CDR последовательность соответствует обозначению Chotia.
3. IL-17-связывающее антитело или его активный фрагмент по п. 1 или 2, представляющее собой человеческое антитело.
4. IL-17-связывающее антитело или его активный фрагмент, которое содержит по меньшей мере один антигенсвязывающий центр, включающий первый домен, имеющий аминокислотную последовательность, представленную в SEQ ID NO:8, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 127, или представленную в SEQ ID NO:10, начиная с аминокислоты в положении 1 и заканчивая аминокислотой в положении 109.
-5-.- -П-р-и-м-е-н-е-н-и-е- -I-L---1-7- -с-в-я-з-ы-ва-ю-щ-е-г-о- -а-н-т-и-т-е-л-а- -и-л-и- -е-г-о- -а-т-и-в-н-о-г-о- -ф-р-а-г-м-е-н-т-а- -п-о- -л-ю-б-о-м-у- -и-з- -п-п-.- -1- --- -4- -д-л-я- -п-о-л-у-ч-е-н-и-я- -л-е-к-а-р-с-т-в-е-н-н-о-г-о- -с-р-е-д-с-т-в-а- -д-л-я- -л-е-ч-е-н-и-я- -о-п-о-с-р-е-д-у-е-м-о-г-о- -I-L-- -1-7- -з-а-б-о-л-е-в-а-н-и-я- -и-л-и- -н-а-р-у-ш-е-н-и-я-.
6. Применение IL-17-связывающего антитела или его активного фрагмента по любому из пп. 1 - 4 для лечения псориаза, псориатического артрита или анкилозирующего спондилита.
-7-.- -Ф-а-р-м-а-ц-е-в-т-и-ч-е-с-к-а-я- -к-о-м-п-о-з-и-ц-и-я-,- -с-о-д-е-р-ж-а-щ-а-я- -I-L---1-7- -с-в-я-з-ы-в-а-ю-щ-е-е- -а-н-т-и-т-е-л-о- -и-л-и- -е-г-о- -а-к-т-и-в-н-ы-й- -ф-р-а-г-м-е-н-т- -п-о- -л-ю-б-о-м-у- -и-з- -п-п-.- -1- --- -4- -в- -с-о-ч-е-т-а-н-и-и- -с- -ф-а-р-м-а-ц-е-в-т-и-ч-е-с-к-и- -п-р-и-е-м-л-е-м-ы-м- -э-к-с-ц-и-п-и-е-н-т-о-м-,- -р-а-з-б-а-в-и-т-е-л-е-м- -и-л-и- -н-о-с-и-т-е-л-е-м-,- -п-р-е-д-н-а-з-н-а-ч-е-н-н-а-я- -д-л-я- -л-е-ч-е-н-и-я- опосредуемого IL-17 -з-а-б-о-л-е-в-а-н-и-я- -и-л-и- -н-а-р-у-ш-е-н-и-я-.
8. Фармацевтическая композиция, содержащая IL-17-связывающее антитело или его активный фрагмент по любому из пп. 1 - 4 в сочетании с фармацевтически приемлемым эксципиентом, разбавителем или носителем, предназначенная для лечения псориаза, псориатического артрита или анкилозиру.
2.3.14. Примеры исчисления срока подачи Заявления и продления исключительного права.
1. Заявка подана - 01.02.2010.
Первое разрешение зарегистрировано - 01.03.2016, т.е. прошло более 5 лет с даты подачи заявки.
Патент выдан - 01.03.2017, т.е. через год после регистрации первого разрешения.
Заявление подано - 01.06.2017.
Анализируются 2 фактора продления:
1) Условие соблюдения срока подачи Заявления.
2) Срок продления исключительного права.
Результаты анализа:
по п. 1) 6-месячный срок подачи Заявления исчисляется либо с даты регистрации 1-го разрешения (01.03.2016), либо с даты выдачи патента (01.03.2017), т.е. с более позднего срока. В данном случае - с даты выдачи патента - 01.03.2017. Таким образом: заявление от 01.06.2017 подано в 6-месячный срок.
Результат соблюдения срока подачи заявления - положительный.
По п. 2) Срок действия исключительного права рассчитывается как разница между регистрацией 1-го разрешения и датой подачи заявки за вычетом 5 лет:
01.03.2016 - 01.02.2010= (6 лет 1 мес.) - 5 лет = 1 год 1 мес.
Таким образом срок действия исключительного права продлен на 1 год и 1 месяц с даты истечения установленного срока действия патента (20 лет):
01.02.2030 + 1 год 1 мес. = 01.03.2031.
Результат продления исключительного права - положительный.
2. Заявка подана - 01.02.2012.
Первое разрешение зарегистрировано - 01.03.2016, т.е. прошло менее 5 лет.
Патент выдан - 01.03.2017, т.е. через год после получения первого разрешения.
Заявление подано - 01.06.2017.
Анализируются 2 фактора продления:
1) Условие соблюдения срока подачи Заявления.
2) Срок продления исключительного права. Результат анализа:
по п. 1) 6-месячный срок подачи Заявления исчисляется либо с даты регистрации 1-го разрешения (01.03.2016), либо с даты выдачи патента (01.03.2017), т.е. с более позднего срока. В данном случае - с даты выдачи патента - 01.03.2017. Таким образом: заявление от 01.06.2017 подано в 6-месячный срок.
Результат соблюдения срока подачи заявления - положительный.
По п. 2) Срок действия исключительного права рассчитывается как разница между регистрацией 1-го разрешения и датой подачи заявки за вычетом 5 лет:
01.03.2016 - 01.02.2012= (4 лет 1 мес.), т.е. менее 5 лет, следовательно срок продления исключительного права не образуется. Дополнительный патент не выдается.
Результат продления исключительного права - отрицательный.
3. Административные процедуры и действия при предоставлении государственной услуги
|
3.1. Прием и регистрация Документов
|
|
|
Заявление может быть подано в окно приема документов Роспатента. При личном обращении в окно приема документов Роспатент, прием и регистрация осуществляется в течение пятнадцати минут.
|
пп. 1 п. 18 Регламента.
пп. 1 п. 52 Регламента
|
|
Заявление может быть представлено через организацию связи. Срок приема и регистрации Документов в этом случае составляет пять рабочих дней с даты поступления Заявления в Роспатент.
|
пп. 2 п. 18 Регламента.
пп. 2, п. 51 Регламента
|
|
Заявление может быть подано с использованием ЕПГУ. Срок приема и регистрации Документов в этом случае составляет пять рабочих дней с даты поступления Заявления в Роспатент.
|
пп. 3 п. 18 Регламента.
п. 43 - 47, пп. 2 п. 51 Регламента
|
|
При приеме Документов проверяется:
1) Наличие Заявления;
2) Заявление должно быть представлено на русском языке. Если Заявление представлено не на русском языке, то проверяется наличие перевода на русский язык заявления и прилагаемых к Заявлению документов.
3) В Заявлении должен быть указан регистрационный номер патента, в отношении которого испрашивается продление срока действия.
4) Возможность прочтения документов. Документы должны поддаваться прочтению.
5) Поданные через ЕПГУ должны соответствовать установленным законодательством РФ требованиям к усиленной квалифицированной электронной подписи (ЭП).
|
п. 16, п. 24 Регламента.
|
|
Если в результате проверки установлено, что одно из вышеперечисленных условий не соблюдено, то Заявителю отказывается в приеме и регистрации Документов и в течение пяти рабочих дней направляется уведомление об отказе в приеме и регистрации заявления. В уведомлении об отказе в приеме и регистрации заявления приводятся основания для отказа, дата поступления непринятых документов и количество листов
|
п. 50, п. 53 Регламента
пп. 2 п. 55, пп. 2 п. 54 Регламента
|
|
В случае отсутствия оснований для отказа осуществляется прием и регистрация Документов. На бланке Заявления проставляется регистрационный номер и дата поступления Заявления или Документов в Роспатент. Сведения о поступивших Документах вводятся в автоматизированную систему делопроизводства.
|
п. 50 Регламента
|
|
Зарегистрированные Документы переводятся в электронный вид и загружаются в соответствующую автоматизированную систему делопроизводства в установленном порядке.
После перевода в электронный вид и размещения документов в автоматизированной системе делопроизводства Документы на бумажном носителе передаются в подразделение, осуществляющее их рассмотрение в срок, не превышающий пять рабочих дней с даты их поступления в Роспатент.
|
пп. 1 п. 54 Регламента
|
|
Принятое и зарегистрированное заявление, непринятое заявление не подлежат возврату заявителю, кроме оригиналов документов, указанных в подпунктах 5, 7 пункта 16 Регламента: регистрационное удостоверение лекарственного препарата, свидетельство о государственной регистрации пестицида или агрохимиката.
|
п. 56 Регламента
|
|
3.2. Проверка соблюдения срока представления заявления и уплаты пошлины
|
|
|
Административная процедура проверки соблюдения срока представления заявления и уплаты пошлины согласно пункту 57 Регламента состоит из следующих административных действий:
- проверка соблюдения срока представления заявления;
- проверка уплаты пошлины.
|
п. 57 Регламента
|
|
3.2.1. Проверка соблюдения срока представления заявления
|
|
|
После поступления Документов в подразделение, осуществляющее их рассмотрение, из подразделения, осуществляющего прием и регистрацию, осуществляется проверка факта представления заявления в сроки, указанные в пункте 3 Порядка. Проверка соблюдения срока представления заявления начинается с даты поступления документов в подразделение, осуществляющее их рассмотрение, и составляет пять рабочих дней.
|
п. 58, 60 Регламента
п. 3 Порядка
|
|
Заявление представляется:
- в период действия патента до истечении шести месяцев со дня получения первого разрешения на применение продукта или с даты выдачи патента в зависимости от того, какой из этих сроков истекает позднее, если с даты подачи заявки на выдачу патента до дня получения первого разрешения на применение прошло более пяти лет;
|
пп. 1 п. 3 Порядка
п. 2 ст. 1363 Кодекса
|
|
- одновременно с ходатайством о восстановлении действия патента, представленным в установленный пунктом 1 статьи 1400 Кодекса срок, или в период рассмотрения ходатайства.
|
пп. 2 п. 3 Порядка
п. 2 ст. 1363 Кодекса
|
|
В случае если заявление представлено в сроки, не соответствующие указанным в абзаце 2 настоящего подпункта, то срок действия исключительного права на изобретение не продлевается. Заявителю направляется уведомление об отказе в продлении срока действия исключительного права на изобретение и удостоверяющего это права патента с указанием основания отказа в течение пяти рабочих дней с даты подписания указанного уведомления.
|
п. 65, пп. 2 п. 66, п. 76 Регламента
|
|
3.2.2. Проверка уплаты пошлины
|
|
|
После поступления Документов в подразделение, осуществляющее их рассмотрение, из подразделения, осуществляющего прием и регистрацию, осуществляется проверка уплаты пошлины. Проверка уплаты пошлины начинается с даты поступления документов в подразделение, осуществляющее их рассмотрение, и составляет десять рабочих дней.
|
п. 57 Регламента
|
|
Проверка уплаты пошлины проводится посредством межведомственного информационного взаимодействия Роспатента с Казначейством России. При проведении проверки используется информация, содержащаяся в ГИС ГМП, в Заявлении и документе, подтверждающем уплату пошлины, если таковой представлен заявителем по собственной инициативе.
|
п. 63 Регламента
|
|
При проверке уплаты пошлины необходимо проверить
следующее:
- уплату пошлины должно осуществлять юридическое или физическое лицо, которое обратилось за осуществлением юридически значимого действия, либо лицо, действующее по его поручению.
- в документе, подтверждающем уплату пошлины, проверяется правильность указания реквизитов банка;
- наличие в графе "назначение платежа" регистрационного номера патента;
- наличие в графе "назначение платежа" наименования юридически значимого действия, за совершение которого уплачена пошлина и соответствующего пункта Приложения к Положению о пошлинах;
- соблюдение 3-х годичного срока действия платежного документа со дня уплаты пошлины;
- соответствие размера уплаченной пошлины установленному размеру.
|
п. 4, 5 Положения о пошлинах
|
|
Патентные пошлины взимаются:
1) за рассмотрение заявления о продлении срока действия исключительного права на изобретение и принятие решения по результатам его рассмотрения - 3000 рублей (подпункт 1.23 приложения N 1 к Положению о пошлинах);
2) за рассмотрение ходатайства о продлении срока представления дополнительных материалов в ответ на запрос с 1 по 6 месяц - 800 рублей за каждый месяц продления (подпункт 1.15.1 приложения N 1 к Положению о пошлинах);
3) за рассмотрение ходатайства о продлении срока представления дополнительных материалов в ответ на запрос с 7 по 10 месяц - 1100 рублей за каждый месяц продления (подпункт 1.15.1 приложения N 1 к Положению о пошлинах);
4) за выдачу дополнительного патент на изобретение - 1500 рублей (подпункт 1.19.2 приложения N 1 к Положению о пошлинах)".
|
п. 29 Регламента
п. 1.15.1, п. 1.19.2, п. 1.23 Положения о пошлинах
|
|
Пошлина за выдачу дополнительного патента уплачивается при уплате пошлины за рассмотрение продления срока действия исключительного права на изобретение. Размер пошлин, предусмотренных подпунктами 1 - 4 пункта 29 Регламента, уменьшается на 30 процентов при обращении за осуществлением юридически значимых действий в электронной форме (пункт 7 Положения о пошлинах)".
|
п. 29, 30 Регламента
п. 7 Положения о пошлинах
|
|
В случае несоответствия сведений, подтверждающих уплату пошлины установленным требованиям, Заявителю направляется уведомление о необходимости представления ходатайства плательщика об уточнении назначении платежа в двух месячный срок с даты направления указанного уведомления.
|
п. 5 Положения о пошлинах
|
|
В случае не подтверждения факта уплаты пошлины или несоответствия установленному размеру, Заявителю направляется уведомляющий документ о начислении с предложением уплатить или доплатить пошлину в двух месячный срок с даты направления указанного уведомления о начислении. В указанном уведомлении сообщается, что если уплата пошлины была произведена ранее, то Заявителю необходимо представить уточненные идентификаторы плательщика и назначение платежа, в соответствии с тем, как они указаны в документе, подтверждающим уплату пошлины.
|
п. 62 Регламента
|
|
По истечении двух месяцев со дня направления уведомления о начислении осуществляется повторная проверка уплаты пошлины в течение десяти рабочих дней со дня истечения двухмесячного срока для уплаты пошлины с даты направления уведомления.
В случае предоставления Заявителем документа подтверждающего уплату пошлины, проверка осуществляется в течение десяти рабочих дней со дня предоставления указанного документа в Роспатент.
|
п. 63 Регламента
|
|
В случае если не уплачены пошлины в размере и порядке, установленных Положением о пошлинах, то срок действия исключительного права на изобретение не продлевается. Заявителю направляется уведомление об отказе в продлении срока действия исключительного права на изобретение и удостоверяющего это права патента с указанием основания отказа в течении пяти рабочих дней со дня окончания проверки уплаты пошлины (в течение пяти рабочих дней с даты подписания указанного уведомления).
|
п. 65, пп. 2 п. 66, п. 76 Регламента
|
|
3.3. Рассмотрение документов
|
|
|
Административная процедура рассмотрения Заявления (Документов) состоит из следующих административных Р действий:
- проверка соответствия представленного заявления и прилагаемых к нему Документов требованиям законодательства Российской Федерации, указанным в пункте 69 Регламента (далее - Требования);
- внесение сведений о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента в Государственный реестр;
- публикация в официальном бюллетене сведений о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента.
|
п. 68 Регламента
|
|
Проверка представленного Заявления и Документов соответствующим Требованиям осуществляется в течение тридцати одного рабочего дня с даты принятия заявления к рассмотрению.
|
п. 69, 72 Регламента
п. 7 Порядка
|
|
Рекомендуется проводить проверку Заявления в следующей последовательности:
1) Наличие принятых в соответствии с законодательством Российской Федерации судебных актов, предусматривающих обеспечительные меры в отношении патента на изобретение, указанного в Заявлении, или в связи с исполнением таких судебных актов. В случае наличия обеспечительных мер заявителю направляется соответствующее уведомление о приостановлении предоставления государственной услуги.
2) Заявление должно быть представлено в отношении одного патента.
|
п. 25 Регламента
|
|
3) Заявление должно быть представлено по Форме <1>.
4) Должен быть представлен оригинал заявления.
5) Заявление должно быть подано от заявителя или его представителя.
6) В случае подачи заявления через патентного поверенного или представителя в Заявлении должны быть указаны сведения о патентном поверенном или представителе.
7) Заявление должно содержать собственноручную подпись поверенного или представителя, с расшифровкой подписи подписавшего лица (фамилия и инициалы). При наличии нескольких правообладателей подписи проставляются всеми правообладателями) или представителя правообладателя(ей) и указывается(ются) расшифровка данной(ых) подписи(ей).
8) В заявлении должен быть указан адрес для переписки, удовлетворяющий требованием быстрой почтовой доставки на территории Российской Федерации.
|
п. 16 Регламента
ст. 1399 Кодекса
п. 5 ст. 1514 Кодекс
пп. 5 п. 2 ст. 1536 Кодекса
Приложение N 1 к Регламенту
|
|
Проверяются условия продления срока действия патента:
1) срок действия патента может быть продлен в соответствии с пунктом 2 статьи 1363 Кодекса;
2) в Государственном реестре изобретений Российской Федерации содержатся сведения о государственной регистрации изобретения, в отношении которого представлено заявление;
3) сведения о патентообладателе, указанные в заявлении, соответствуют сведениям, содержащимся в Государственном реестре, относящимся к изобретению, в отношении которого представлено заявление;
4) лицо, представившее заявление, обладает полномочиями на ведение дел с Роспатентом;
5) правовая охрана изобретения, в отношении которого представлено заявление, не прекращена или не признана недействительной в установленном законодательством Российской Федерации порядке;
6) соблюден установленный пунктом 1 статьи 1400 Кодекса срок представления ходатайствао восстановлении действия патента, если заявление представляется одновременно с ходатайством или в период рассмотрения ходатайства;
7) формула изобретения, содержащая совокупность признаков запатентованного изобретения (далее - формула изобретения), характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено в установленном законодательством Российской Федерации порядке разрешение (далее - разрешение).
|
п. 7, п. 8 Порядка
|
|
Представленная формула изобретения соответствует условиям продления срока действия патента, предусмотренным пунктом 8 Порядка:
1) устанавливается, характеризует ли формула изобретения продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение.
Формула изобретения характеризует продукт, который относится к лекарственному средству, пестициду или агрохимикату и на применение которого получено разрешение, если:
- в формуле изобретения продукт охарактеризован в виде соединения или группы соединений, описываемых общей структурной формулой, и из описания изобретения следует возможность его (ее) использования в качестве активного ингредиента лекарственного средства, пестицида или агрохимиката, при этом совокупность признаков, определяющих объем правовой охраны продукта, характеризующих соединение или группу соединений, описываемых общей структурной формулой, идентична активному ингредиенту лекарственного средства, пестицида или агрохимиката, указанного в полученном разрешении на применение этого продукта, а описание изобретения содержит информацию о том, что соединение или группа соединений, описываемых общей структурной формулой, обладает такой активностью, которая позволяет его (ее) использовать в указанном в разрешении лекарственном средстве, пестициде или агрохимикате;
- в формуле изобретения продукт охарактеризован в виде композиции лекарственного средства, пестицида или агрохимиката определенного назначения, совокупность признаков, определяющих объем правовой охраны продукта, характеризующих композицию, указанную в формуле изобретения, идентична характеристике композиции лекарственного средства, пестицида или агрохимиката, указанной в разрешении (назначением, составом, формой, если она приведена в формуле изобретения или следует из состава композиции);
2) устанавливается, относится ли полученное разрешение к первому разрешению, и определяется день получения первого разрешения:
- для лекарственных средств - в соответствии с положениями Федерального закона от 12 апреля 2010 года N 61-ФЗ "Об обращении лекарственных средств" (Собрание законодательства Российской Федерации, 2010, N 16, ст. 1815; N 31, ст. 4161; N 42, ст. 5293; N 49, ст. 6409; 2011, N 50, ст. 7351; 2012, N 26, ст. 3446; N 53, ст. 7587; 2013, N 27, ст. 3477; N 48, ст. 6165; 2014, N 11, ст. 1098; N 43, ст. 5797; N 52, ст. 7540; 2015, N 10, ст. 1404; N 27, ст. 3951; N 29, ст. 4359, 4367) первым разрешением на применение изобретения, относящегося к лекарственному средству, является регистрационное удостоверение лекарственного препарата, выдаваемое со сроком действия пять лет, а днем получения первого разрешения является дата регистрации лекарственного средства, указанная в регистрационном удостоверении лекарственного препарата;
- для пестицидов или агрохимикатов - в соответствии с положениями Федерального закона от 19 июля 1997 года N 109-ФЗ "О безопасном обращении с пестицидами и агрохимикатами" (Собрание законодательства Российской Федерации, 1997, N 29, ст. 3510; 2003, N 2, ст. 153, 167; 2004, N 27, ст. 2711; 2006, N 43, ст. 4412; 2008, N 26, ст. 3022; 2009, N 1, ст. 17, 21; 2010, N 41, ст. 5189; 2011, N 30, ст. 4590, 4596; 2015, N 15, ст. 4359) первым разрешением на применение изобретения, относящегося к пестициду или агрохимикату, является свидетельство о государственной регистрации пестицида или агрохимиката, выдаваемого на срок два года или десять лет, а днем получения первого разрешения является дата регистрации свидетельства о государственной регистрации пестицида или агрохимиката.
|
|
|
Если Документы не соответствуют установленным Требованиям и условия продления не соблюдены, направляется запрос с указанием оснований, по которым продление срока исключительного права на изобретение и удостоверяющего это право патента не может быть осуществлено, и предложением в течение трех месяцев со дня направления указанного запроса представить запрашиваемые и (или) надлежаще оформленные документы (далее - запрос о представлении Документов).
|
п. 70 Регламента
|
|
В запросе о представлении Документов заявитель информируется о том, что в случае непредставления дополнительных и (или) надлежаще оформленных документов в трехмесячный срок с даты направления заявителю будет направлено уведомление об отказе в продлении срока действия исключительного права на изобретение и удостоверяющего это право патента.
|
п. 71 Регламента
|
|
Срок повторной проверки соответствия Документов Требованиям составляет двадцать один рабочий день с даты поступления ответа на указанный запрос.
|
п. 72 Регламента
|
|
Срок представления запрашиваемых материалов может быть продлен не более чем на десять месяцев по ходатайству патентообладателя, представленному в течение трех месяцев с даты направления запроса недостающих и/или правильно оформленных документов либо дополнительных материалов, которые необходимы для продления срока действия патента на изобретение.
Срок рассмотрения ходатайства о продлении срока ответа на запрос составляет месячный срок с даты поступления указанного ходатайства в Роспатент.
|
п. 9 Порядка
|
|
В случае непредставления ответа на запрос по истечении трех месяцев с даты его направления, Заявителю направляется уведомление об отказе в продлении срока действия исключительного права на изобретение и удостоверяющего это право патента в течение пяти рабочих дней со дня истечения трехмесячного срока для представления запрошенных документов (с даты подписания указанного уведомления об отказе).
|
п. 78, пп. 2 п. 79 Регламента
|
|
В случае удовлетворения Документов Требованиям и соблюдении условий продления, заявителю направляется уведомление о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента в течение пяти рабочих дней с даты подписания указанного уведомления.
|
пп. 1 п. 79 Регламента
|
|
3.4. Внесение сведений о продлении срока действия действия исключительного права на изобретение и удостоверяющего это право патента в Государственный реестр
|
|
|
В случае удовлетворения Документов Требованиям и соблюдения условий продления, сведения о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента вносятся в Государственный реестр изобретений.
|
пп. 2 п. 73 Регламента
|
|
Максимальный срок внесения сведений в Государственный реестр изобретений один рабочий день с даты принятия решения о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента (с даты подписания уведомления о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента).
|
п. 4 Регламента
|
|
В Государственный реестр вносятся следующие сведения с приведением перед ними в скобках соответствующих международных цифровых кодов для идентификации библиографических данных (кодов ИНИД), если таковые предусмотрены стандартом ВОИС:
- регистрационный номер дополнительного патента;
- регистрационный номер патента;
- номер и дата регистрации первого разрешения на применение продукта;
- дата подачи заявления о продлении срока действия патента;
- срок действия исключительного права на изобретение по дополнительному патенту;
- дата публикации сведений о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента;
- номер официального бюллетеня;
- основание для продления срока действия исключительного права на изобретение и удостоверяющего это право патента (заявление с указанием даты его поступления);
- дата внесения записи о продлении срока действия патента в Государственный реестр.
|
п. 73 Регламента
|
|
3.5. Публикация в официальном бюллетене сведений о продлении срока действия исключительного права на изобретение и удостоверяющего это право патента
|
|
|
Административное действие осуществляется, если соблюдены условия продления, и заключается в публикации в официальном бюллетене сведений о продлении срока действия исключительного права на изобретение и выдаче дополнительного патента с указанием в нем сведений, предусмотренных пунктом 11 Порядка, за исключением сведений о продлении срока действия исключительного права на секретное изобретение.
|
п. 75 Регламента
|
|
Максимальный срок осуществления указанного административного действия двадцать три рабочих дня с даты направления (выдачи) заявителю уведомления о продлении срока действия исключительного права на изобретение.
|
п. 76 Регламента
|
|
В случае если срок действия патента продлевается, заявителю выдается (направляется по адресу для переписки, указанному в заявлении дополнительный патент, содержащий следующие сведения:
- наименование или фамилия, имя, отчество (последнее - при наличии) патентообладателя;
- регистрационный номер дополнительного патента;
- регистрационный номер патента;
- номер и дата регистрации первого разрешения на применение продукта;
- дата подачи заявления;
- срок действия исключительного права на изобретение по дополнительному патенту;
- дата внесения записи о продлении срока действия патента в Государственный реестр;
- формула изобретения, содержащая совокупность признаков запатентованного изобретения, характеризующих продукт, на применение которого получено разрешение.
|
п. 11 Порядка
|
--------------------------------
<1> Форма - форма заявления о продлении срока действия исключительного права на изобретение, представленная в приложении N 1 к Регламенту.