См. Документы Министерства сельского хозяйства Российской Федерации
МИНИСТЕРСТВО СЕЛЬСКОГО ХОЗЯЙСТВА РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРИКАЗ
от 6 марта 2018 г. N 101
ОБ УТВЕРЖДЕНИИ ПРАВИЛ
ПРОВЕДЕНИЯ ДОКЛИНИЧЕСКОГО ИССЛЕДОВАНИЯ ЛЕКАРСТВЕННОГО
СРЕДСТВА ДЛЯ ВЕТЕРИНАРНОГО ПРИМЕНЕНИЯ, КЛИНИЧЕСКОГО
ИССЛЕДОВАНИЯ ЛЕКАРСТВЕННОГО ПРЕПАРАТА ДЛЯ ВЕТЕРИНАРНОГО
ПРИМЕНЕНИЯ, ИССЛЕДОВАНИЯ БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННОГО
ПРЕПАРАТА ДЛЯ ВЕТЕРИНАРНОГО ПРИМЕНЕНИЯ
В целях реализации статьи 12 Федерального закона от 12 апреля 2010 г. N 61-ФЗ "Об обращении лекарственных средств" (Собрание законодательства Российской Федерации, 2010, N 16, ст. 1815; N 31, ст. 4161; N 42, ст. 5293; N 49, ст. 6409; 2011, N 50, ст. 7351; 2012, N 26, ст. 3446; N 53, ст. 7587; 2013, N 27, ст. 3477; N 48, ст. 6165; 2014, N 11, ст. 1098; N 43, ст. 5797; N 52, ст. 7540; 2015, N 10, ст. 1404; N 27, ст. 3951; N 29, ст. 4359, ст. 4367, ст. 4388; N 51, ст. 7245; 2016, N 1, ст. 9; N 23, ст. 3287; N 27, ст. 4194, ст. 4238, ст. 4283; 2017, N 31, ст. 4791, ст. 4827; 2018, N 1, ст. 9) и в соответствии с подпунктом 5.2.25(29) пункта 5 Положения о Министерстве сельского хозяйства Российской Федерации, утвержденного постановлением Правительства Российской Федерации от 12 июня 2008 г. N 450 (Собрание законодательства Российской Федерации, 2008, N 25, ст. 2983; N 32, ст. 3791; N 42, ст. 4825; N 46, ст. 5337; 2009, N 1, ст. 150; N 3, ст. 378; N 6, ст. 738; N 9, ст. 1119, ст. 1121; N 27, ст. 3364; N 33, ст. 4088; 2010, N 4, ст. 394; N 5, ст. 538; N 16, ст. 1917; N 23, ст. 2833; N 26, ст. 3350; N 31, ст. 4251, ст. 4262; N 32, ст. 4330; N 40, ст. 5068; 2011, N 6, ст. 888; N 7, ст. 983; N 12, ст. 1652; N 14, ст. 1935; N 18, ст. 2649; N 22, ст. 3179; N 36, ст. 5154; 2012, N 28, ст. 3900; N 32, ст. 4561; N 37, ст. 5001; 2013, N 10, ст. 1038; N 29, ст. 3969; N 33, ст. 4386; N 45, ст. 5822; 2014, N 4, ст. 382; N 10, ст. 1035; N 12, ст. 1297; N 28, ст. 4068; 2015, N 2, ст. 491; N 11, ст. 1611; N 26, ст. 3900; N 35, ст. 4981; N 38, ст. 5297; N 47, ст. 6603; 2016, N 2, ст. 325; N 28, ст. 4741; N 33, ст. 5188; N 35, ст. 5349; N 47, ст. 6650; N 49, ст. 6909, ст. 6910; 2017, N 26, ст. 3852; N 51, ст. 7824), приказываю:
1. Утвердить прилагаемые Правила проведения доклинического исследования лекарственного средства для ветеринарного применения, клинического исследования лекарственного препарата для ветеринарного применения, исследования биоэквивалентности лекарственного препарата для ветеринарного применения (далее - Правила).
2. Установить, что Правила применяются к исследованиям, проводимым в рамках доклинических исследований лекарственных средств для ветеринарного применения и клинических исследований лекарственных препаратов для ветеринарного применения, планы проведения которых утверждены после вступления в силу настоящего приказа.
3. Настоящий приказ вступает в силу по истечении шести месяцев со дня его официального опубликования.
Министр
А.Н.ТКАЧЕВ
Утверждены
приказом Минсельхоза России
от 6 марта 2018 г. N 101
ПРАВИЛА
ПРОВЕДЕНИЯ ДОКЛИНИЧЕСКОГО ИССЛЕДОВАНИЯ ЛЕКАРСТВЕННОГО
СРЕДСТВА ДЛЯ ВЕТЕРИНАРНОГО ПРИМЕНЕНИЯ, КЛИНИЧЕСКОГО
ИССЛЕДОВАНИЯ ЛЕКАРСТВЕННОГО ПРЕПАРАТА ДЛЯ ВЕТЕРИНАРНОГО
ПРИМЕНЕНИЯ, ИССЛЕДОВАНИЯ БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННОГО
ПРЕПАРАТА ДЛЯ ВЕТЕРИНАРНОГО ПРИМЕНЕНИЯ
I. Общие требования
1. Настоящие Правила устанавливают порядок проведения доклинического исследования лекарственного средства для ветеринарного применения (далее - доклиническое исследование), клинического исследования лекарственного препарата для ветеринарного применения (далее - клиническое исследование), исследования биоэквивалентности лекарственного препарата для ветеринарного применения (далее - исследование биоэквивалентности).
2. Доклиническое исследование, клиническое исследование (в том числе исследование биоэквивалентности) проводятся по утвержденному разработчиком лекарственного средства (препарата) для ветеринарного применения (далее - разработчик) плану с ведением протоколов этих исследований и составлением отчетов, в которых содержатся результаты этих исследований.
3. Для организации и проведения доклинического исследования, клинического исследования разработчик может привлекать организации, имеющие необходимую материально-техническую базу и квалифицированных специалистов в соответствующей области исследования (далее - сторонняя организация).
4. Полномочия, ответственность и обязанности лиц, ответственных за проведение доклинического, клинического исследований, устанавливаются приказом (распоряжением) руководителя разработчика.
Полномочия, ответственность и обязанности лиц, участвующих в проведении доклинического, клинического исследований, а также лиц, ответственных за осуществление исследований, проводимых в рамках доклинического, клинического исследований, устанавливаются приказом (распоряжением) руководителя разработчика, либо в случае привлечения сторонней организации - приказом (распоряжением) руководителя сторонней организации, копия которого направляется разработчику в течение трех рабочих дней с момента его подписания.
5. Лица, ответственные за проведение доклинического, клинического исследований, а также лица, ответственные за осуществление исследований, проводимых в рамках доклинического, клинического исследований, должны быть ознакомлены разработчиком с результатами ранее проведенных исследований и с информацией, которая может оказать влияние на результаты исследования.
6. Отчеты о результатах доклинического и клинического исследований составляются разработчиком с учетом заключений сторонних организаций (в случае привлечения таких организаций).
7. Документы, оформляемые при проведении доклинического и клинического исследований в соответствии с настоящими Правилами, подлежат учету в электронном и (или) бумажном виде организацией, их оформившей, в журнале (журналах) учета. В журнале (журналах) учета должны быть указаны наименование, дата, номер документа (при наличии), а также фамилия, имя, отчество (при наличии) лица, осуществившего запись в журнале (журналах) учета.
8. Документы, оформляемые при проведении доклинического и клинического исследований в соответствии с настоящими Правилами, или их копии подлежат постоянному хранению у разработчика.
9. Документы, оформляемые сторонней организацией при проведении доклинического и клинического исследований в соответствии с настоящими Правилами, или их копии подлежат хранению в сторонних организациях (в случае их привлечения) в течение трех лет. Необходимость дальнейшего хранения в сторонних организациях указанных документов или их копий определяется договором, заключенным разработчиком и сторонней организацией.
II. Правила проведения доклинического исследования
10. К доклиническим исследованиям относятся:
изучение фармакодинамики действующего вещества (действующих веществ) лекарственного средства для ветеринарного применения (далее - лекарственное средство), включающее изучение специфической (фармакологической) активности лекарственного средства;
изучение общетоксических свойств лекарственного средства (в том числе острой, субхронической и (или) хронической токсичности, местнораздражающего действия лекарственного средства);
изучение специфической токсичности лекарственного средства (в том числе иммунотоксичности, аллергенности, репродуктивной токсичности, тератогенности, эмбриотоксичности, мутагенности с прогнозом канцерогенности);
изучение переносимости здоровыми животными тех видов, которым предназначается исследуемый лекарственный препарат для ветеринарного применения (далее - целевые животные), при однократном и многократном введении в терапевтической (профилактической) и повышенных дозах;
изучение фармакокинетики действующего вещества (действующих веществ) лекарственного средства и (или) его метаболита (метаболитов) в организме целевых животных;
изучение сроков выведения остаточных количеств действующего вещества (действующих веществ) и (или) его метаболита (метаболитов) из организма целевых продуктивных животных в целях обеспечения безопасности продукции животного происхождения после применения соответствующего лекарственного препарата для ветеринарного применения (далее - лекарственный препарат) в максимальной рекомендованной разработчиком дозе.
11. В случае, если действующим веществом исследуемого лекарственного средства является действующее вещество, входящее в состав другого зарегистрированного в Российской Федерации лекарственного препарата, к отчету о результатах доклинического исследования взамен отчетов о результатах исследования фармакодинамики действующего вещества лекарственного средства, а также специфической токсичности лекарственного средства при наличии научного обоснования может прилагаться обзор научных работ по указанным видам исследований.
12. Сбор, обработка и хранение информации, получаемой в ходе доклинического исследования, должны обеспечивать получение достоверного и обоснованного представления о безопасности и фармакологической активности лекарственного средства.
13. Лица, ответственные за проведение доклинического исследования, и лица, ответственные за осуществление исследований, проводимых в рамках доклинического исследования, должны иметь высшее ветеринарное, медицинское, фармацевтическое, химическое, биотехнологическое или биологическое образование и стаж работы в области доклинических исследований.
14. Лица, участвующие в проведении доклинического исследования, должны иметь квалификацию в соответствующей области исследования.
15. Лица, ответственные за проведение доклинического исследования, лица, ответственные за проведение исследований, проводимых в рамках доклинического исследования, и лица, участвующие в проведении доклинического исследования, должны быть ознакомлены под роспись руководителем организации, проводящей исследование, с планом исследования, полномочиями, обязанностями при проведении доклинического исследования, информацией об исследуемом лекарственном средстве, а также с информацией о возможных опасностях, возникающих при работе с лекарственным средством.
16. Лица, ответственные за проведение доклинического исследования, должны:
не допускать отклонений от плана исследования, внесения в него изменений без согласия разработчика, за исключением изменений, необходимых для устранения угрозы жизни и здоровью людей и необоснованной гибели животных, используемых при проведении доклинического исследования (далее - экспериментальные животные);
обеспечить своевременный сбор полученных результатов, регистрацию отклонений от плана исследования с указанием причин и оценкой влияния внесенных изменений на полученные результаты, а также при необходимости принять меры по устранению выявленных отклонений;
обеспечить интерпретацию и анализ получаемых результатов, подготовку отчета о результатах доклинического исследования, конфиденциальность полученных результатов.
17. Руководитель организации, проводящей исследование, должен обеспечивать выполнение требований, установленных планом исследования, объективность и независимость проведения исследования и нести ответственность за достоверность получаемых результатов.
18. Помещения, предназначенные для проведения доклинического исследования, должны соответствовать требованиям законодательства Российской Федерации и обеспечивать необходимые для проведения доклинического исследования температурные и влажностные режимы.
19. Планировка помещений, предназначенных для проведения доклинического исследования, должна обеспечивать:
изоляцию (карантин) поступающих экспериментальных животных, больных экспериментальных животных и экспериментальных животных, подозреваемых в носительстве инфекций;
хранение кормов, оборудования и инвентаря для ухода за экспериментальными животными изолированно от мест содержания экспериментальных животных;
изолированное хранение образцов исследуемых лекарственных средств, в том числе архивных образцов лекарственных средств;
изолированное хранение реактивов, реагентов, тест-систем и стандартных веществ;
изолированное хранение получаемых отходов до их утилизации или уничтожения;
изолированное хранение документов;
возможность вскрытия павших экспериментальных животных;
проведение мойки, дезинфекции инвентаря, подготовку кормов и удаление отходов.
20. Возможность несанкционированного доступа посторонних лиц в помещения, предназначенные для проведения доклинического исследования, должна быть исключена.
21. Помещения, предназначенные для проведения доклинического исследования, должны в соответствии с графиком, установленным руководителем организации, проводящей доклиническое исследование, подвергаться влажной уборке с использованием дезинфицирующих средств, обеспечивающей безопасность работы в них, но не оказывающей влияния на получаемые результаты.
22. При проведении доклинического исследования должно применяться измерительное и испытательное оборудование (далее - оборудование), которое прошло поверку или иной необходимый контроль.
23. Оборудование должно эксплуатироваться и обслуживаться в соответствии с документацией на оборудование, утвержденной производителем оборудования (далее - документация).
24. Сотрудникам организации, проводящей исследования, эксплуатирующим оборудование или обеспечивающим его обслуживание, должны быть доступны следующие сведения:
наименование оборудования, производителя;
модель оборудования, серийный (заводской) номер (при наличии), дата получения и постановки на учет, дата запуска в эксплуатацию, инвентарный номер;
место расположения оборудования;
сведения о месте хранения документации на оборудование;
данные о сотруднике, ответственном за использование оборудования;
данные о сотруднике, ответственном за техническое обслуживание оборудования;
записи о плановом обслуживании оборудования и результатах проведения профилактических осмотров, с указанием даты и подписи сотрудника, ответственного за техническое обслуживание оборудования;
записи о повреждениях, отказах в работе, ремонте оборудования, с указанием даты и подписи сотрудника, ответственного за техническое обслуживание оборудования.
25. Используемые при проведении доклинического исследования реактивы и реагенты, стандартные вещества и тест-системы должны отвечать задачам исследования, соответствовать требованиям, указанным в плане исследования, применяться до истечения срока их годности, иметь маркировку, позволяющую их идентифицировать.
26. Хранение реактивов, реагентов, тест-систем и стандартных веществ должно осуществляться в условиях, обеспечивающих их стабильность в процессе хранения и установленных производителем, в упаковке, обеспечивающей защиту от загрязнения или порчи, в отдельной зоне помещений, предназначенных для проведения доклинического исследования, с ограниченным доступом. Параметры окружающей среды зоны хранения реактивов, реагентов, тест-систем и стандартных веществ должны систематически регистрироваться в порядке, утвержденном организацией, проводящей доклиническое исследование.
27. Все процедуры, связанные с уходом за экспериментальными животными, подлежат учету на бумажном носителе и (или) в электронном виде.
28. Вновь поступившие экспериментальные животные подлежат карантинированию для оценки состояния здоровья.
29. На экспериментальных животных, поступивших в организацию, проводящую доклиническое исследование, должны быть оформлены ветеринарные сопроводительные документы в случаях, предусмотренных законодательством Российской Федерации.
30. Больные экспериментальные животные и экспериментальные животные, подозреваемые в носительстве инфекций, должны быть изолированы от здоровых экспериментальных животных и, при необходимости, подвергнуты лечению. Все процедуры, связанные с диагностикой заболевания, назначенным лечением и его результатами, а также оценка влияния заболевания и проведенного лечения на получаемые результаты подлежат учету на бумажном носителе и (или) в электронном виде.
31. Для обеспечения индивидуального наблюдения в процессе проведения доклинического исследования экспериментальные животные должны быть идентифицированы. В отношении мелких грызунов допускается групповая идентификация.
32. Все клетки, вольеры, контейнеры, предназначенные для содержания экспериментальных животных, должны быть промаркированы.
33. Экспериментальные животные, используемые при проведении доклинического исследования одного лекарственного средства, должны содержаться изолированно от экспериментальных животных, используемых при проведении доклинического исследования другого лекарственного средства.
34. Различные виды экспериментальных животных должны содержаться изолированно в отдельных клетках, вольерах, контейнерах.
35. Экспериментальные животные одного вида должны содержаться в одинаковых условиях, оптимальных для данного вида животных, иметь свободный доступ к кормам и воде. Корма и вода должны быть безопасными для здоровья экспериментальных животных, обеспечивать потребности животных в питательных веществах и не должны влиять на результаты исследования.
36. Экспериментальные животные в течение не менее чем 30 суток (птицы в течение 15 суток) до начала исследований и в период исследований не должны получать никаких других лекарственных препаратов, если иное не предусмотрено планом исследования. При проведении доклинического исследования лекарственного средства, обладающего пролонгированным действием, указанный период должен быть увеличен.
37. При работе с экспериментальными животными следует руководствоваться следующими принципами:
а) использование вида (видов) экспериментальных животных, выбор которого (которых) научно обоснован и соответствует поставленным задачам исследования;
б) использование экспериментальных животных в минимальном количестве, которое требуется для получения научно достоверных и статистически обоснованных результатов;
в) использование, при возможности, научно обоснованных альтернативных методов и материалов, включающих беспозвоночных животных, культуры клеток, микроорганизмы взамен теплокровных экспериментальных животных;
г) использование экспериментальных животных таким образом, чтобы свести к минимуму причиняемые им неудобства и боль;
д) проведение продолжительных, болезненных манипуляций, хирургических операций на экспериментальных животных только с применением седативных, анальгетических лекарственных препаратов, лекарственных препаратов для наркоза;
е) умерщвление безболезненным способом в конце или в процессе доклинического исследования экспериментальных животных, которые будут испытывать сильные или постоянные боли, физические страдания, постоянную функциональную недостаточность, неподдающиеся устранению, а также экспериментальных животных при невозможности их дальнейшего использования.
Экспериментальные животные на начало проведения доклинического исследования должны быть здоровыми и не являться носителями агентов, способных повлиять на результаты исследования, если иное не предусмотрено планом доклинического исследования.
38. Отбор проб биологических материалов проводится в индивидуальные пробирки (флаконы, контейнеры), которые должны маркироваться с указанием идентификационных данных экспериментального животного (группы животных), времени отбора пробы биологического материала и наименования исследуемого лекарственного средства или кодироваться в соответствии с планом исследования.
39. Если иное не предусмотрено планом исследования, пробы биологического материала должны замораживаться и храниться в замороженном виде до их исследования.
40. Сотрудником, проводившим отбор проб биологического материала, должен составляться документ с указанием даты отбора, идентификационных данных экспериментального животного (группы животных), их пола, возраста, массы. Пробы биологического материала должны передаваться в лабораторию с указанным документом для исследования.
41. Объем пробы биологического материала, необходимый для исследования, должен определяться исходя из метода исследования.
42. При проведении доклинического исследования должны использоваться образцы лекарственного средства:
в той же лекарственной форме, которая планируется для применения;
произведенные по технологии, которая предусматривается для включения в промышленный регламент или в иной документ, определяющий технологию производства;
соответствующие требованиям качества, предусмотренным проектом нормативного документа на лекарственное средство;
с читаемой маркировкой, включающей наименование образца лекарственного средства или присвоенный кодовый номер, наименование и количественное содержание действующего вещества (действующих веществ) (или активности), номер серии, условия хранения и срок годности.
43. В случае если содержание действующего вещества в лекарственной форме при проведении доклинических исследований не на целевых животных не позволяет установить дозу, вызывающую функциональные изменения в организме животных, допускается при наличии научного обоснования использование лекарственной формы с большей концентрацией действующего вещества, чем планируется для применения на целевых животных.
44. Образцы лекарственного средства должны сопровождаться представленной разработчиком документацией, содержащей указания об условиях и сроках хранения, информацию о мерах по обеспечению безопасности работы с исследуемым лекарственным средством, растворителями и при необходимости информацией о процедуре растворения, устройствами для введения лекарственного средства экспериментальным животным.
45. Образцы исследуемых лекарственных средств подлежат учету при приеме, расходе, возврате или утилизации в соответствии с процедурой, утвержденной организацией, проводящей исследование.
46. Хранение образцов исследуемого лекарственного средства должно осуществляться в условиях, установленных разработчиком, в упаковке, обеспечивающей защиту от загрязнения или порчи, обеспечивающих их стабильность в процессе хранения, в отдельной зоне помещений, предназначенных для проведения доклинического исследования, с ограниченным доступом. Параметры окружающей среды зоны хранения образцов должны регулярно регистрироваться в порядке, утвержденном организацией, проводящей исследование.
47. Образцы лекарственного средства, представленные для проведения доклинического исследования, должны иметь срок годности, достаточный для завершения доклинического исследования. Использование в доклиническом исследовании образцов лекарственного средства с истекшим сроком годности или хранившихся в условиях, не соответствующих условиям хранения, установленным разработчиком, не допускается. В случае длительного доклинического исследования, превышающего срок годности лекарственного средства, условия замены образцов лекарственного средства и критерии приемлемости должны быть описаны в плане исследования.
48. При подготовке образцов лекарственного средства для введения экспериментальным животным не должна допускаться их контаминация другими лекарственными средствами и инфекционными агентами, должна быть обеспечена безопасность лиц, участвующих в проведении исследования, и окружающей среды.
49. Введение лекарственных средств должно осуществляться таким образом, чтобы каждое экспериментальное животное гарантированно получило определенную дозу лекарственного средства.
50. Уничтожение остатков исследуемого лекарственного средства осуществляется в соответствии со статьей 59 Федерального закона от 12 апреля 2010 г. N 61-ФЗ "Об обращении лекарственных средств" (далее - Федеральный закон N 61-ФЗ).
51. Организация, проводящая исследование, должна утвердить стандартные операционные процедуры, в которых должен быть подробно и последовательно описан порядок осуществления и учета всех лабораторных и производственных операций, включая:
а) поступление, идентификацию, маркировку, обработку, отбор проб, использование, хранение и уничтожение (утилизацию) исследуемых образцов лекарственных средств, стандартных образцов и тест-систем (в случае их использования);
б) обслуживание и поверку оборудования;
в) приготовление реактивов, питательных сред;
г) ведение записей, отчетов, протоколов и их хранение;
д) обслуживание помещений, в которых проводится доклиническое исследование;
е) прием, транспортировку, размещение, описание, идентификацию экспериментальных животных, уход за ними.
52. Разработчик должен утвердить план каждого исследования, проводимого в рамках доклинического исследования, с указанием даты его утверждения. Указанный план исследования должен содержать:
наименование исследования, проводимого в рамках доклинического исследования;
наименование и адрес организации, проводящей исследование, место проведения исследования;
фамилию, имя, отчество (при наличии) лица, ответственного за проведение исследования, и лиц, участвующих в проведении исследований;
цель исследования;
задачу исследования;
срок (месяц, год) начала и планируемый срок (месяц, год) окончания исследования;
сведения об исследуемом лекарственном средстве (физические, химические, фармацевтические, биологические свойства);
сведения о стандартном образце (образцах) (в случае его (их) использования);
принципы использования видов экспериментальных животных, а также критерии выбора используемых в исследовании видов экспериментальных животных, их возраста, пола, массы тела, критерии включения (исключения) экспериментальных животных, порядка их замены;
методы исследования с обоснованием их выбора;
схему исследования и ее обоснование;
количество экспериментальных животных в группе, способа (способов) и пути (путей) введения экспериментальным животным исследуемого лекарственного средства, кратности применения и продолжительности фазы исследования на экспериментальных животных и аналитической фазы, периодичности оценки состояния экспериментальных животных и отбора проб, оцениваемые показатели в процессе исследования и методики оценки, их обоснование;
описание биологического материала, отбираемого для проведения исследования, способов его отбора и хранения, их обоснование;
описание процедуры статистической обработки результатов исследования;
обоснование необходимости (отсутствия необходимости) проведения валидации метода (методов) исследования;
критерии оценки контролируемых в процессе исследования показателей;
порядок внесения изменений в план исследования;
ссылки на литературные источники (в случае их использования);
дополнительную информацию (в случае наличия).
53. План исследования составляется разработчиком или сторонней организацией (в случае ее привлечения для проведения исследования).
54. План исследования подписывает лицо, ответственное за проведение доклинического исследования, с указанием должности, места работы.
55. При проведении исследования лица, участвующие в проведении исследования, должны вести протокол исследования на бумажном носителе и (или) в электронном виде, в котором должны фиксироваться действия, предусмотренные планом исследования.
56. Протокол исследования должен включать:
наименование исследования, проводимого в рамках доклинического исследования;
описание использованного оборудования, средств измерения и реактивов, реагентов, стандартных образцов и тест-систем (в случае их использования);
первичные данные о результатах измерений и наблюдений (в том числе хроматограммы и фотографии при их наличии);
результаты вычислений и преобразования данных (в том числе промежуточные);
описание и оценку процедур статистического анализа с указанием использованного программного обеспечения;
сведения об используемых экспериментальных животных (вид, возраст, количество, масса, пол и количество групп экспериментальных животных в каждом виде исследований), условия содержания и кормления экспериментальных животных, параметры окружающей среды в помещениях, в которых содержатся экспериментальные животные;
сведения, имеющие непосредственное отношение к исследованию и позволяющие воспроизвести ход исследования.
Для автоматически регистрируемых параметров с большим числом индивидуальных значений допускается представление вместо первичных данных о результатах измерений и наблюдений результатов первичной обработки этих данных.
57. Протокол исследования должен быть подписан всеми лицами, участвовавшими в проведении исследования, с указанием фамилии, имени, отчества (при наличии), ученой степени (при наличии), а также с указанием даты подписания и номера протокола исследования, позволяющих идентифицировать данный протокол.
58. Содержание протокола должно позволять однозначно идентифицировать исследование, использовавшиеся образцы лекарственного средства, вид исследования, методы, лиц, участвующих в получении данных и в подготовке проведения исследования, измерительное и испытательное оборудование, реагенты и реактивы, стандартные образцы и тест-системы (в случае их использования).
59. Изменения сведений, содержащихся в протоколе исследования, оформляются в виде дополнений к указанному протоколу, которые должны быть подписаны всеми лицами, участвовавшими в проведении исследования, с указанием причин изменений, даты и номера дополнения к протоколу исследования.
60. После завершения исследования, проведенного в рамках доклинического исследования, лицом, ответственным за проведение данного исследования, составляется и подписывается отчет о результатах исследования, который должен содержать:
наименование вида исследования;
дату и номер, позволяющие идентифицировать данный отчет;
наименование, адрес организации, проводившей исследование, и место проведения исследования;
даты начала и завершения исследования;
цель и задачи исследования;
фамилию, имя, отчество (при наличии), ученую степень (при наличии) лица, ответственного за проведение данного вида исследования, и лиц, участвующих в проведении исследования;
описание исследуемого лекарственного средства, включая состав, физико-химические, биологические, фармацевтические свойства, номер серии, срок годности;
описание хода исследования с указанием использованных материалов и методов;
описание использованных оборудования, средств измерения и реактивов, реагентов, стандартных образцов и тест-систем (в случае их использования);
информацию об экспериментальных животных (вид, пол, возраст, масса тела, количество животных в группе);
способ введения, дозы и кратность введения лекарственного средства;
описание и оценку процедур статистического анализа с указанием использованного программного обеспечения;
результаты исследования со ссылками на соответствующие первичные данные о результатах измерений и наблюдений, а также их статистический анализ;
выводы из проведенного исследования.
61. В случае отклонения от плана исследования лицом, ответственным за проведение исследования, проводимого в рамках доклинического исследования, должны составляться изменения в план исследования с указанием причин отклонения, которые утверждаются разработчиком и прилагаются к плану исследования.
62. В случае привлечения для проведения доклинического исследования сторонней организации после завершения исследования на основании указанного в пункте 60 настоящих Правил отчета о результатах исследования лицом, ответственным за проведение данного вида исследования, должно составляться и подписываться заключение, содержащее выводы из проведенного исследования.
63. После завершения всех исследований, проведенных в рамках доклинического исследования, разработчиком составляется отчет о результатах доклинического исследования на основании указанных в пункте 60 настоящих Правил отчетов о результатах исследований и, при наличии, заключений, указанных в пункте 62 настоящих Правил.
64. Отчет о результатах доклинического исследования должен быть подписан руководителем разработчика.
65. Отчет о результатах доклинического исследования должен содержать:
наименование отчета;
наименование лекарственного средства;
наименования, адреса разработчика, производителя лекарственного средства и сторонних организаций (в случае привлечения таких организаций для проведения доклинического исследования);
даты начала и окончания доклинического исследования;
дату и номер отчета, позволяющие его идентифицировать;
цель и задачи доклинического исследования;
краткое описание проведенного доклинического исследования, информацию о стандартном образце (при наличии), продолжительность доклинического исследования, информацию об экспериментальных животных, результаты доклинического исследования;
оглавление, включая перечень приложений, таблиц;
перечень сокращений и определение терминов, используемых в отчете;
описание исследуемого лекарственного средства, включая состав, физико-химические, биологические, фармацевтические свойства;
виды исследований, проведенных в рамках доклинического исследования;
вид, возраст, количество, массу и пол экспериментальных животных, использованных в каждом виде исследований, проведенных в рамках доклинического исследования, условия их содержания;
количество групп экспериментальных животных в каждом виде исследований, проведенных в рамках доклинического исследования;
режим дозирования, кратность и способ введения исследуемого лекарственного средства;
описание и оценку процедур статистического анализа с указанием использованного программного обеспечения;
обобщенные результаты исследований со ссылками на отчеты о результатах каждого вида исследований, проведенных в рамках доклинического исследования, а также анализ указанных результатов;
выводы о безопасности, качестве и эффективности лекарственного средства, в том числе о сроках его выведения из организма животных и безопасности продукции животного происхождения после применения соответствующего лекарственного препарата. Указанные выводы должны содержать сведения о фармакологической активности, фармакокинетике, фармакодинамике и возможности последующего проведения клинического исследования, степени опасности (безопасности) лекарственного средства для животных, которым предназначается исследуемое лекарственное средство, безопасности продукции животного происхождения, полученной от таких животных.
66. К отчету о результатах доклинического исследования должны прилагаться планы исследований (или их копии, заверенные разработчиком лекарственного средства), отчеты о результатах каждого вида исследований, проведенных в рамках доклинического исследования, и при наличии заключения сторонних организаций (или копии заключений и отчетов о результатах каждого вида исследований, проведенных в рамках доклинического исследования, заверенные этими организациями).
67. Если в процессе исследования использовались методы контроля, подлежащие валидации, копии протоколов валидации должны прилагаться к отчету о результатах доклинического исследования.
III. Правила проведения клинического исследования
68. Возможность проведения клинического исследования должна основываться на результатах доклинического исследования.
69. Переносимость лекарственных препаратов здоровыми животными устанавливается на основании данных, полученных при проведении доклинического исследования на целевых животных.
70. До начала клинического исследования разработчиком должна быть проведена оценка соотношения возможного риска с прогнозируемой пользой для целевых животных. Оценка эффективности лекарственного препарата должна быть начата и продолжена только в случае преобладания прогнозируемой пользы над риском.
71. В случае осуществления исследования, проводимого в рамках клинического исследования в Российской Федерации, разработчик должен уведомить Россельхознадзор в электронной форме или на бумажном носителе о дате начала и месте осуществления указанного исследования в течение 10 рабочих дней с момента утверждения плана исследования.
72. Россельхознадзор в течение 10 рабочих дней с момента поступления уведомления разработчика о дате начала и месте осуществления исследования, проводимого в рамках клинического исследования, должен сообщить разработчику в электронной форме или на бумажном носителе о получении указанного уведомления.
73. Процедуры сбора, обработки и хранения информации, получаемой в ходе клинического исследования, должны обеспечивать получение достоверного и обоснованного представления о безопасности и эффективности лекарственного препарата.
74. В случае обнаружения опасности и (или) возникновения угрозы для здоровья животных и (или) людей и (или) загрязнения окружающей среды, лицо, ответственное за осуществление исследования, проводимого в рамках клинического исследования, должно немедленно приостановить исследование, проводимое в рамках клинического исследования, до момента установления причины возникновения опасности и (или) возникновения угрозы для здоровья животных и (или) людей и (или) загрязнения окружающей среды и проинформировать разработчика о приостановлении исследования, проводимого в рамках клинического исследования, на бумажном носителе или в электронной форме.
75. Решение о прекращении исследования, проводимого в рамках клинического исследования, принимается разработчиком в случае подтверждения того, что применение лекарственного препарата представляет угрозу для здоровья животных и (или) людей и (или) загрязнения окружающей среды.
76. Решение о возобновлении исследования, проводимого в рамках клинического исследования, принимается разработчиком в случае подтверждения того, что применение лекарственного препарата не представляет угрозу для здоровья животных и (или) людей и (или) загрязнения окружающей среды.
77. Разработчик должен сообщить в Россельхознадзор в электронной форме или на бумажном носителе о прекращении или о возобновлении исследования, проводимого в рамках клинического исследования, в срок, не превышающий 5 рабочих дней со дня принятия решения о прекращении или о возобновлении клинического исследования.
78. Лица, участвующие в проведении клинического исследования, должны иметь квалификацию в соответствующей области исследования.
79. Лицо, ответственное за проведение клинического исследования, и лица, ответственные за осуществление исследований, проводимых в рамках клинического исследования, должны иметь высшее ветеринарное образование и соответствующие целям клинических исследований подготовку и стаж работы в области ветеринарии не менее трех лет.
80. Лицо, ответственное за проведение клинического исследования, лица, ответственные за осуществление исследований, проводимых в рамках клинического исследования, и лица, участвующие в проведении клинического исследования, должны быть ознакомлены под роспись руководителем организации, проводящей исследование, с планом исследования, полномочиями, обязанностями при осуществлении исследования, информацией об исследуемом лекарственном препарате, в том числе с информацией о потенциальных рисках, возникающих при работе с лекарственным препаратом.
81. Лица, ответственные за проведение клинического исследования, должны:
не допускать отклонений от плана исследования, внесения в него изменений без согласия разработчика, за исключением изменений, необходимых для устранения угрозы жизни и здоровью людей и необоснованной гибели целевых животных;
обеспечить своевременный сбор полученных результатов, регистрацию отклонений от плана исследования с указанием причин и оценкой влияния внесенных изменений на полученные результаты, а также при необходимости принять меры по устранению выявленных отклонений;
обеспечить интерпретацию и анализ получаемых результатов, подготовку отчета о результатах клинического исследования, конфиденциальность полученных результатов.
82. Руководитель организации, проводящей исследование, должен обеспечить выполнение требований, установленных планом исследования, объективность и независимость осуществления исследования и несет ответственность за достоверность получаемых результатов.
83. Помещения, предназначенные для проведения клинического исследования, должны соответствовать требованиям законодательства Российской Федерации и обеспечивать необходимые для проведения клинического исследования температурные и влажностные режимы.
84. Планировка помещений, предназначенных для проведения клинического исследования, должна обеспечивать:
изоляцию (карантин) поступающих целевых животных, больных целевых животных и целевых животных, подозреваемых в носительстве инфекций;
раздельное содержание целевых животных, используемых в разных видах клинических исследований;
хранение кормов, оборудования и инвентаря для ухода за целевыми животными изолированно от мест содержания целевых животных;
изолированное хранение образцов исследуемых лекарственных препаратов, в том числе архивных образцов лекарственных препаратов;
изолированное хранение реактивов, реагентов, тест-систем и стандартных образцов;
изолированное хранение получаемых отходов до их утилизации или уничтожения;
изолированное хранение документов;
возможность вскрытия павших целевых животных;
проведение мойки, дезинфекции инвентаря, подготовки кормов и удаление отходов.
85. При проведении клинического исследования должно применяться измерительное и испытательное оборудование, реактивы и реагенты, стандартные образцы и тест-системы, соответствующие требованиям, указанным в пунктах 22 - 26 настоящих Правил.
86. Клинические исследования должны проводиться на каждом виде целевых животных, каждой возрастной группе (возрастных группах), для которого (которых) предполагается использовать лекарственный препарат.
87. В клиническом исследовании должны использоваться целевые животные в количестве, достаточном для обеспечения статистической значимости исследования с учетом возможности исключения целевых животных из исследования и их замены.
88. После предварительного отбора целевых животных проводится их клиническое обследование, включающее осмотр и, при необходимости, лабораторные исследования, с целью допуска для использования в исследовании целевых животных, соответствующих целям и задачам исследования.
89. На целевых животных, поступивших в организацию, проводящую клиническое исследование, должны быть оформлены ветеринарные сопроводительные документы в случаях, предусмотренных законодательством Российской Федерации.
90. Для оценки состояния здоровья, необходимого для использования целевых животных в исследовании, лицо, ответственное за осуществление исследования, должно принять решение о необходимости карантинирования целевых животных и о сроках их карантинирования.
91. Для обеспечения индивидуального наблюдения в процессе выполнения исследования целевые животные должны быть идентифицированы. В отношении целевых животных, используемых при проведении клинических исследований лекарственных препаратов, предназначенных для групповых обработок, допускается групповая идентификация.
92. В период исследования целевые сельскохозяйственные животные без крайней необходимости не должны перемещаться из одного помещения в другое.
93. Целевые животные должны иметь свободный доступ к воде. Корма и вода должны быть безопасными для здоровья целевых животных, обеспечивать потребности целевых животных в питательных веществах и не должны влиять на результаты исследования.
94. При проведении клинического исследования необходимо руководствоваться принципами, указанными в подпунктах "а", "б", "г", "д" пункта 37 настоящих Правил.
95. Собственник используемого в исследовании животного должен быть проинформирован об указанном исследовании и связанных с ним рисках, в том числе о целесообразности гуманного умерщвления животного в случае, если в процессе клинического исследования его физическое состояние резко ухудшилось и дальнейшее проведение клинического исследования причиняет животному сильные или постоянные боли, физические страдания, постоянную функциональную недостаточность, неподдающиеся устранению.
96. Информирование собственника животного об исследовании, в котором используется животное, и связанных с ним рисках осуществляется в письменной форме до начала осуществления исследования, а о наступлении рисков, указанных в пункте 95 настоящих Правил, - не позднее трех часов с момента их наступления.
97. Отбор проб биологических материалов должен проводиться в индивидуальные пробирки (флаконы, контейнеры), которые должны маркироваться с указанием идентификационных данных целевого животного (группы животных), времени отбора пробы биологического материала и наименования исследуемого лекарственного препарата или кодироваться в соответствии с планом исследования.
98. Если иное не предусмотрено планом исследования, пробы биологического материала должны замораживаться и храниться в замороженном виде до их исследования.
99. Сотрудником, проводившим отбор проб биологического материала, должен составляться документ с указанием даты отбора, идентификационных данных целевого животного (группы животных), их пола, возраста, массы. Пробы биологического материала передаются в лабораторию с указанным документом для исследования.
100. Объем пробы биологического материала, необходимый для исследования, определяется исходя из метода исследования.
101. При проведении клинического исследования должны использоваться образцы лекарственного препарата:
в той же лекарственной форме, которая планируется для применения;
произведенные по технологии, которая предусматривается для включения в промышленный регламент или в иной документ, определяющий технологию производства;
соответствующие требованиям качества, предусмотренным проектом нормативного документа на лекарственный препарат;
с маркировкой, соответствующей требованиям статьи 46 Федерального закона N 61-ФЗ.
102. Образцы лекарственного препарата должны сопровождаться представленной разработчиком документацией, содержащей указания об условиях и сроках хранения, информацию о мерах по обеспечению безопасности работы с исследуемым лекарственным препаратом, растворителями и информацией о процедуре растворения, информацией о комплектности, а также, если необходимо, устройствами для введения лекарственного препарата целевым животным.
103. Образцы исследуемого лекарственного препарата подлежат учету при приеме, расходе, возврате или утилизации в соответствии с процедурой, утвержденной организацией, проводящей исследование.
104. Хранение образцов исследуемого лекарственного препарата должно осуществляться в условиях, установленных разработчиком, в упаковке, обеспечивающей защиту от загрязнения или порчи, в отдельной зоне помещений, предназначенных для проведения клинического исследования, с ограниченным доступом.
105. Образцы лекарственного препарата, предоставленные для проведения клинического исследования, должны иметь срок годности, достаточный для завершения клинического исследования. Использование в клиническом исследовании образцов лекарственного препарата с истекшим сроком годности или хранившихся в условиях, не соответствующих условиям хранения, установленным разработчиком, не допускается. В случае длительного клинического исследования, превышающего срок годности лекарственного средства, условия замены образцов лекарственного препарата и критерии приемлемости должны быть описаны в плане исследования.
106. При подготовке образцов лекарственного препарата для введения целевым животным не должна допускаться их контаминация другими лекарственными средствами и инфекционными агентами, должна быть обеспечена безопасность для лиц, участвующих в осуществлении исследования, и окружающей среды.
107. Уничтожение остатков исследуемого лекарственного препарата осуществляется в соответствии со статьей 59 Федерального закона N 61-ФЗ.
108. Организация, проводящая исследование, должна утвердить стандартные операционные процедуры, в которых подробно и последовательно должен быть описан порядок осуществления и учета всех лабораторных и производственных операций, включая:
а) прием, транспортировку, размещение, идентификацию, обработку и кормление целевых животных;
б) обслуживание и поверку оборудования;
в) приготовление реактивов, питательных сред;
г) ведение записей, отчетов, протоколов и их хранение;
д) обслуживание помещений, в которых проводится клиническое исследование;
е) приготовление кормов для целевых животных;
ж) поступление, идентификацию, маркировку, обработку, отбор проб, использование, хранение и уничтожение (утилизацию) исследуемых образцов лекарственных препаратов и стандартных образцов (в случае их использования).
109. Разработчик должен утвердить план каждого исследования, проводимого в рамках клинического исследования, с указанием даты его утверждения. Указанный план исследования должен содержать:
наименование исследования, проводимого в рамках клинического исследования;
наименование и адрес организации, проводящей исследование, место осуществления исследования;
фамилию, имя, отчество (при наличии) лица, ответственного за осуществление исследования, и лиц, участвующих в осуществлении исследований;
цель исследования;
задачи исследования;
обоснование плана исследования, проводимого в рамках клинического исследования;
срок (месяц, год) начала и планируемый срок (месяц, год) окончания исследования;
сведения об исследуемом лекарственном препарате (наименование, качественный и количественный состав, номер серии, объем серии, дата производства, срок годности, физические, химические, биологические, фармацевтические свойства);
сведения о препарате сравнения (наименование, качественный и количественный состав действующих веществ, качественный состав вспомогательных веществ, номер серии, срок годности, физические, химические, биологические, фармацевтические свойства, номер регистрационного удостоверения лекарственного препарата) в случае его использования при проведении клинического исследования;
обоснование выбора референтного препарата (при исследовании биоэквивалентности);
описание и обоснование используемых в исследованиях методов, оборудования и средств измерения;
критерии выбора используемых в исследовании целевых животных, их вида, возраста, пола, массы тела, критерии включения (исключения) целевых животных, порядка их замены;
схему исследования и ее обоснование;
количество целевых животных в группе, периодичность оценки состояния целевых животных и отбора проб, оцениваемые в процессе исследования показатели и методики их оценки, их обоснование;
описание биологического материала, отбираемого для осуществления исследования, способов его отбора и хранения, их обоснование;
методы распределения целевых животных по группам, информация о предшествующей и сопутствующей терапии;
критерии постановки диагноза и способы его подтверждения;
описание лабораторных исследований, проводимых в процессе исследования;
способы, пути введения, доза и режим дозирования исследуемого лекарственного препарата и (при наличии) препарата сравнения, их обоснование;
критерии оценки эффективности и безопасности исследуемого лекарственного препарата;
порядок внесения изменений в план вида исследования;
описание процедуры статистической обработки результатов исследования;
ссылки на литературные источники (в случае их использования); дополнительную информацию (в случае наличия).
110. План исследования должен составляться разработчиком или сторонней организацией (в случае ее привлечения для осуществления исследования).
111. План исследования подписывает лицо, ответственное за проведение клинического исследования, с указанием должности, места работы.
112. Лица, участвующие в осуществлении исследования, должны вести протокол исследования на бумажном носителе и (или) в электронном виде, в котором фиксируются действия, предусмотренные планом исследования.
113. Протокол исследования должен включать:
наименование исследования, проводимого в рамках клинического исследования;
описание использованного оборудования, средств измерения и реактивов, реагентов, стандартных образцов и тест-систем (при использовании);
первичные данные о результатах измерений и наблюдений (при возможности индивидуальной идентификации животных);
описание и оценку процедур статистического анализа с указанием использованного программного обеспечения (дополнительно при исследовании биоэквивалентности указываются все значения индивидуальных концентраций и фармакокинетических параметров, а также данные описательной статистики, включая геометрическое среднее, медиану, арифметическое среднее, стандартное отклонение, коэффициент вариации, максимальные и минимальные значения для каждого из сравниваемых лекарственных препаратов, а также индивидуальные фармакокинетические кривые или график средней фармакокинетической кривой с доверительными интервалами на линейной или логарифмической шкалах);
сведения об используемых целевых животных (вид, возраст, количество, масса, пол и количество групп целевых животных), условия содержания и кормления целевых животных, параметры окружающей среды в помещениях, в которых содержатся целевые животные;
сведения о возникновении нежелательных реакций и серьезных нежелательных реакций и мерах по их устранению;
сведения, имеющие непосредственное отношение к исследованию и позволяющие воспроизвести ход исследования;
дизайн исследования, в том числе способ введения лекарственных препаратов, дозу и режим дозирования, интервал времени между введением лекарственных препаратов, биологический материал, схему отбора проб биологического материала, условия их хранения (при исследовании биоэквивалентности);
описание аналитического метода (при исследовании биоэквивалентности);
хроматограммы (при исследовании биоэквивалентности хроматографическим или масспектрометрическим методами);
описание методов фармакокинетического анализа (при исследовании биоэквивалентности);
описание критериев биоэквивалентности (при исследовании биоэквивалентности).
Для автоматически регистрируемых параметров с большим числом индивидуальных значений допускается представление вместо первичных данных о результатах измерений и наблюдений результатов первичной обработки этих данных.
114. Протокол исследования должен быть подписан всеми лицами, участвовавшими в осуществлении исследования, с указанием фамилии, имени, отчества (при наличии), ученой степени (при наличии), а также с указанием даты подписания и номера протокола исследования, позволяющих идентифицировать данный протокол.
115. Содержание протокола должно позволять однозначно идентифицировать исследование, использовавшиеся образцы лекарственного препарата, вид исследования, методы, лиц, участвующих в получении данных и в подготовке осуществления исследования, а также оборудование, реагенты и реактивы, стандартные образцы и тест-системы (в случае их использования).
116. Изменения сведений, содержащихся в протоколе исследования, оформляются в виде дополнений к указанному протоколу, которые подписываются всеми лицами, участвовавшими в проведении доклинического исследования, с указанием причин изменений, даты и номера дополнения к протоколу исследования.
117. После завершения исследования, проведенного в рамках клинического исследования, лицом, ответственным за осуществление данного исследования, должен быть составлен и подписан отчет о результатах исследования, который должен содержать:
наименование вида исследования;
дату и номер, позволяющие идентифицировать данный отчет;
наименование, адрес организации, проводившей исследование, и место осуществления исследования;
даты начала и завершения исследования;
цель и задачи исследования;
фамилию, имя, отчество (при наличии), ученую степень (при наличии), место работы лица, ответственного за осуществление данного вида исследования, и лиц, участвующих в осуществлении исследования;
описание исследуемого лекарственного препарата и препарата сравнения (при наличии), включая состав, физико-химические, биологические, фармацевтические свойства, номер серии, срок годности;
описание хода исследования с указанием использованных материалов и методов;
описание использованного оборудования, средств измерения и реактивов, реагентов, препаратов сравнения и тест-систем (в случае их использования);
режим дозирования, кратность и путь введения исследуемого лекарственного препарата;
вид, возраст, пол, количество целевых животных в каждой группе, условия кормления;
результаты исследования со ссылками на соответствующие первичные данные о результатах измерений и наблюдений, их статистический анализ;
выводы из проведенного исследования.
118. При исследовании биоэквивалентности отчет о результатах исследования должен содержать сведения, указанные в пункте 117 настоящих Правил, а также следующую информацию:
обоснование выбора метода определения действующего вещества, или метаболита, или изомера, обладающих фармакологической активностью (далее - аналит) в биологическом материале;
описание использованного способа расчета фармакокинетических параметров;
результаты определения содержания аналита в биологическом материале, полученные для каждого целевого животного, соответствующие средние значения и величины стандартных отклонений;
статистические критерии, использованные для оценки биоэквивалентности, и результаты этой оценки. Для каждого сравниваемого лекарственного препарата должны быть указаны все значения индивидуальных концентраций и фармакокинетических параметров, наряду с данными описательной статистики, включая геометрическое среднее, медиану, арифметическое среднее, стандартное отклонение, коэффициент вариации, максимальные и минимальные значения. Индивидуальные кривые "концентрация-время" следует представлять на линейной или логарифмической шкалах. Должен быть описан метод получения фармакокинетических параметров из исходных данных и количество точек в терминальной логарифмической фазе, использованных для оценки константы скорости терминальной элиминации.
119. В случае отклонения от плана исследования лицом, ответственным за осуществление исследования, проводимого в рамках клинического исследования, составляются изменения в план исследования с указанием причин отклонения, которые утверждаются разработчиком и прилагаются к плану исследования.
120. В случае привлечения для проведения клинического исследования сторонней организации после завершения исследования на основании указанного в пункте 117 настоящих Правил отчета о результатах исследования лицом, ответственным за осуществление данного вида исследования, составляется и подписывается заключение, содержащее выводы из проведенного исследования.
121. После завершения всех исследований, проведенных в рамках клинического исследования, разработчиком составляется отчет о результатах клинического исследования на основании указанных в пункте 117 настоящих Правил отчетов о результатах исследований и, при наличии, заключений, указанных в пункте 120 настоящих Правил.
122. Отчет о результатах клинического исследования должен быть подписан руководителем разработчика.
123. Отчет о результатах клинического исследования должен содержать:
наименование отчета;
наименование лекарственного препарата;
наименования, адреса разработчика, производителя лекарственного препарата и сторонних организаций (в случае привлечения таких организаций для проведения клинического исследования);
даты начала и окончания клинического исследования;
дату и номер отчета, позволяющие его идентифицировать;
цели и задачи клинического исследования;
краткое описание проведенного клинического исследования, информацию о препарате сравнения (при наличии), продолжительность клинического исследования, дозирование лекарственного препарата, виды целевых животных, результаты клинического исследования;
оглавление, включая перечень приложений, таблиц;
перечень сокращений и определение терминов, используемых в отчете;
краткое описание проведенного доклинического исследования, информацию о стандартном образце (при наличии), продолжительность доклинического исследования, информацию об экспериментальных животных, результаты доклинического исследования;
описание исследуемого лекарственного препарата, включая состав, физико-химические, биологические, фармацевтические свойства;
виды исследований, проведенных в рамках клинического исследования;
сведения о целевых животных и распределении их по группам (вид, возраст, количество, масса и пол целевых животных, использованных в каждом виде исследований, условия их содержания, кормления и поения, количество групп целевых животных в каждом виде исследований);
дозу (дозы), режим дозирования, способы введения и применения, время приема лекарственного препарата, продолжительность лечения (далее - схема применения лекарственного препарата);
описание и оценку процедур статистического анализа с указанием использованного программного обеспечения;
обобщенные результаты исследований со ссылками на отчеты о результатах каждого вида исследований, проведенных в рамках клинического исследования, а также анализ указанных результатов, включая описание нежелательных реакций (краткое описание нежелательных реакций, их анализ, списки нежелательных реакций, которые наблюдались у целевых животных), описание случаев гибели целевых животных и других серьезных нежелательных реакций, оценку клинико-лабораторных показателей животных (при наличии), параметры жизненно важных функций организма целевых животных и другую информацию по вопросам безопасности, а также оценку эффективности и безопасности лекарственного препарата;
анализ литературных данных (при наличии);
выводы о переносимости лекарственного препарата целевыми животными, об установлении оптимальных дозировок лекарственного препарата и курса лечения на конкретной группе животных с определенным заболеванием (схемы применения лекарственного препарата), безопасности и эффективности лекарственного препарата, предназначенного для лечения определенных заболеваний животных, или эффективности лекарственного препарата для профилактики заболеваний животных, возможностях расширения показаний к применению зарегистрированного лекарственного препарата и выявления ранее неизвестных побочных действий.
124. К отчету о результатах клинического исследования должны прилагаться планы (или их копии, заверенные разработчиком лекарственного препарата), отчеты о результатах каждого вида исследований, проведенных в рамках клинического исследования, и, при наличии, заключения сторонних организаций (или копии заключений и отчетов о результатах каждого вида исследований, проведенных в рамках клинического исследования, заверенные этими организациями).
125. При исследовании биоэквивалентности к отчету о результатах клинического исследования должна прилагаться копия протокола валидации метода определения аналита, содержащего сведения о линейности в необходимом диапазоне концентраций, влиянии биологического материала на получаемый результат исследования, пределе обнаружения метода, пределе количественного определения метода, селективности метода, точности метода, прецизионности метода, стабильности аналита в биологическом материале, а также в случае определения аналита хроматографическим или масспектрометрическим методами - хроматограммы, подтверждающие представленные результаты валидации.
IV. Правила исследования биоэквивалентности
126. Исследование биоэквивалентности является видом клинического исследования, проведение которого осуществляется для определения скорости всасывания и выведения одного или нескольких обладающих фармакологической активностью действующих веществ и (или) их метаболитов, количества лекарственного препарата, достигающего системного кровотока, и результаты которого позволяют сделать вывод о биоэквивалентности воспроизведенного лекарственного препарата в определенных лекарственной форме и дозировке, соответствующих лекарственной форме и дозировке референтного лекарственного препарата.
127. Два лекарственных препарата, содержащих одинаковое количество действующего вещества, считаются биоэквивалентными, если они являются фармацевтически эквивалентными и их биодоступность (по скорости и степени, с которой действующее вещество или активная часть молекулы действующего вещества абсорбируются из лекарственного препарата и становятся доступными в месте своего действия) после применения в одинаковой молярной дозе укладывается в допустимые пределы. Указанные пределы устанавливаются для обеспечения сопоставимости биофармацевтических свойств лекарственной формы, в которой выпускаются лекарственные препараты, in vivo (то есть сопоставимости их по эффективности и безопасности).
128. Исследования биоэквивалентности не проводятся в отношении:
а) иммунобиологических лекарственных препаратов;
б) лекарственных препаратов растительного происхождения, в отношении которых не был установлен состав;
в) лекарственных препаратов, не достигающих системного кровотока;
г) лекарственных препаратов, предназначенных для интрацистернального или внутриматочного введения;
д) пробиотических лекарственных препаратов.
129. Действующее вещество в референтном и в воспроизведенном лекарственных препаратах должно иметь одинаковую молекулярную форму.
130. Перед началом исследования биоэквивалентности должен быть проведен анализ образцов воспроизведенного и референтного лекарственных препаратов на содержание и подлинность действующего вещества (действующих веществ).
131. Дозы референтного и воспроизведенного лекарственных препаратов должны быть одинаковыми. При выборе дозы в пересчете на действующее вещество следует руководствоваться инструкцией по ветеринарному применению референтного лекарственного препарата.
132. В случае если референтный лекарственный препарат может применяться в разных дозах, следует использовать максимальную рекомендованную инструкцией по ветеринарному применению референтного лекарственного препарата дозу (далее - рекомендованная доза).
В случае если представлено научное обоснование нецелесообразности использования в исследовании биоэквивалентности лекарственного препарата максимальной рекомендованной дозы, допускается использовать любую дозу из диапазона доз, указанных в инструкции по ветеринарному применению референтного лекарственного препарата.
133. В случае если при введении рекомендованной дозы действующее вещество (действующие вещества) в крови не достигает (не достигают) доступных для измерения современными методами значений, допускается использование более высоких доз, но не более трехкратной рекомендованной дозы при условии, что использование таких доз не приводит к возникновению нежелательных реакций организма животного.
134. Подготовку воспроизведенного и референтного лекарственных препаратов к введению целевым животным следует проводить одинаковым способом, так, чтобы это не оказало влияния на результаты исследования. Введение лекарственных препаратов должно осуществляться таким образом, чтобы каждое животное гарантированно получило определенную дозу лекарственного препарата. Для лекарственных препаратов в форме таблеток с риской допускается использование разделенной по риске части таблетки, в том случае, если нормативным документом на лекарственный препарат предусмотрен контроль однородности дозирования частей таблеток.
135. Если референтный лекарственный препарат выпускается с разной дозировкой, исследование биоэквивалентности воспроизведенного лекарственного препарата должно проводиться по одной из дозировок при соблюдении следующих условий:
качественный состав действующих веществ и вспомогательных веществ референтного лекарственного препарата с разной дозировкой одинаков (за исключением красителей и ароматизаторов);
соотношение между содержанием действующих веществ и вспомогательных веществ референтного лекарственного препарата с разной дозировкой одинаково;
технология производства референтного лекарственного препарата с разной дозировкой одинакова.
136. Многократное введение при исследовании биоэквивалентности должно применяться для:
лекарственных препаратов с замедленным высвобождением действующего вещества, предназначенных для многократного введения и имеющих тенденцию к накоплению в организме;
лекарственных препаратов с насыщаемым выведением;
лекарственных препаратов, концентрацию которых в крови существующими аналитическими методами не представляется возможным определить при однократном введении.
В указанных случаях при выборе кратности введения следует руководствоваться инструкцией по ветеринарному применению референтного лекарственного препарата и определять концентрацию действующего вещества в крови перед введением следующей дозы лекарственного препарата.
137. При отсутствии в плане исследования обоснования для многократного введения при исследовании биоэквивалентности применяется однократное введение лекарственного препарата.
138. В случае если инструкцией по ветеринарному применению референтного лекарственного препарата предусмотрено несколько путей введения, исследование биоэквивалентности воспроизведенного лекарственного препарата может проводиться любым из указанных путей введения. При этом в плане исследования должно быть указано обоснование взаимозаменяемости разных путей введения.
В иных случаях должно проводиться исследование биоэквивалентности при всех способах введения, предусмотренных проектом инструкции по ветеринарному применению воспроизведенного лекарственного препарата.
139. Дизайн исследования биоэквивалентности может быть перекрестным, параллельным или последовательным.
140. Перекрестный дизайн предусматривает введение первой группе целевых животных референтного лекарственного препарата, второй группе целевых животных - воспроизведенного лекарственного препарата. После определенного периода времени первой группе животных вводится воспроизведенный лекарственный препарат, второй группе животных - референтный лекарственный препарат.
Период между введениями референтного лекарственного препарата и воспроизведенного лекарственного препарата группе животных должен быть не менее чем шестикратный период полувыведения действующего (действующих) вещества (веществ) и (или) его (их) метаболитов. При наличии научного обоснования продолжительность указанного периода может быть изменена, но должна обеспечивать максимально полное выведение действующего (действующих) вещества (веществ) и (или) его (их) метаболитов из организма животного, а также отсутствие остаточных физиологических эффектов от введения лекарственных препаратов.
При исследовании биоэквивалентности лекарственных препаратов, действующие вещества которых являются аналогами веществ, образующихся (присутствующих) в организме животного в процессе его жизнедеятельности (далее - эндогенные вещества), продолжительность периода между введениями референтного лекарственного препарата и воспроизведенного лекарственного препарата группе животных должна быть обоснована. Базовые уровни эндогенного вещества в крови животных первой и второй группы, определенные перед введением препарата, не должны оказывать влияния на результаты исследования.
При использовании перекрестного дизайна каждому целевому животному следует вводить лекарственный препарат в одной и той же дозе во все периоды исследования.
Перекрестный дизайн следует использовать в случае невозможности применения последовательного или параллельного дизайна на имеющемся количестве животных либо при изучении биоэквивалентности лекарственных препаратов, действующие вещества которых являются эндогенными веществами.
Перекрестный дизайн не должен использоваться при проведении исследования биоэквивалентности на тех видах целевых животных, физиологическое состояние организма которых значительно изменится за период проведения исследования биоэквивалентности, за исключением случаев, когда есть научное обоснование возможности использования перекрестного дизайна.
141. Параллельный дизайн предусматривает одновременное введение первой группе животных референтного лекарственного препарата и второй группе животных воспроизведенного лекарственного препарата.
Параллельный дизайн следует использовать для проведения исследования биоэквивалентности в следующих случаях:
действующее вещество или его метаболиты вызывают физиологические изменения в организме животного, которые впоследствии могут повлиять на фармакокинетику данного вещества при повторном введении;
действующее вещество или его метаболиты имеют длительный период полувыведения или тенденцию к кумуляции в организме животного, в связи с чем повышается риск присутствия остаточных количеств лекарственных препаратов после введения лекарственных препаратов либо изменяется физиологическое состояние животных;
общий объем крови в организме животного исключает возможность повторного использования таких животных в исследовании.
142. Последовательный дизайн предусматривает последовательное введение группе животных референтного лекарственного препарата, а затем воспроизведенного лекарственного препарата.
Период между введениями референтного лекарственного препарата и воспроизведенного лекарственного препарата группе животных должен быть не менее чем шестикратный период полувыведения действующего (действующих) вещества (веществ) и (или) его (их) метаболитов. При наличии научного обоснования продолжительность указанного периода может быть изменена, но должна обеспечивать максимально полное выведение действующего (действующих) вещества (веществ) и (или) его (их) метаболитов из организма животного, а также отсутствие остаточных физиологических эффектов от введения лекарственных препаратов.
Последовательный дизайн не должен использоваться при проведении исследования биоэквивалентности на тех видах целевых животных, физиологическое состояние организма которых значительно изменится за период проведения исследования биоэквивалентности.
143. В случае если инструкцией по ветеринарному применению референтного лекарственного препарата предусмотрено применение референтного лекарственного препарата нескольким видам животных, исследование биоэквивалентности воспроизведенного лекарственного препарата возможно на одном из указанных видов животных, если они относятся к теплокровным животным.
144. Для проведения исследования биоэквивалентности могут использоваться животные любого пола. Возраст животных, порода, масса, уровень продуктивности (для сельскохозяйственных животных) в группе животных, которой вводится референтный лекарственный препарат, и группе животных, которой вводится воспроизведенный лекарственный препарат, должны быть аналогичными. Масса не должна выходить за пределы 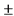 20% для животных массой до 20 кг и
20% для животных массой до 20 кг и 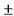 10% для животных массой более 20 кг от средней массы по группе животных.
10% для животных массой более 20 кг от средней массы по группе животных.
145. В день введения лекарственного препарата доступ животных к корму должен быть ограничен, если иное не обосновано в плане исследования.
146. Животные для проведения исследования биоэквивалентности в течение не менее чем 30 суток (птицы в течение 15 суток) до начала исследования биоэквивалентности и в период исследования биоэквивалентности не должны получать лекарственных препаратов, если иное не предусмотрено планом исследования. При проведении исследования биоэквивалентности лекарственного препарата, обладающего пролонгированным действием, указанный период должен быть увеличен.
147. Число животных в одной группе должно быть не менее шести. Большее число животных может потребоваться для сравнения референтного и воспроизведенного лекарственных препаратов, обладающих значительной вариабельностью фармакокинетических параметров, а также при проведении исследования биоэквивалентности на животных, у которых не представляется возможным проводить многократный забор биологического материала в необходимом количестве.
148. В качестве биологического материала для исследования биоэквивалентности используются сыворотка, плазма крови или цельная кровь. В случае невозможности определения концентрации действующего вещества (действующих веществ) и (или) его (их) метаболитов в плазме крови, сыворотке, цельной крови, допускается определение действующего вещества (действующих веществ) и (или) его (их) метаболитов в тканях.
149. Схема и продолжительность отбора проб биологического материала определяется формой фармакокинетической кривой (графика зависимости концентрации действующего вещества в отобранном биологическом материале от времени, прошедшего с момента введения лекарственного препарата). Выбор времени отбора проб биологического материала должен обеспечивать получение не менее трех точек для фазы первоначального возрастания концентрации действующего вещества и не менее пяти точек для фазы ее снижения, за исключением лекарственных препаратов для внутривенного введения.
В отношении лекарственных препаратов для внутривенного введения выбор времени отбора проб биологического материала должен обеспечивать получение не менее пяти точек для фазы снижения концентрации действующего вещества.
В отношении лекарственных препаратов с пролонгированным действием выбор времени отбора проб биологического материала должен дополнительно обеспечивать получение не менее трех точек для фазы, когда максимальная концентрация действующего вещества (действующих веществ) относительно постоянна.
150. Для получения достоверного значения максимальной концентрации действующего вещества (далее - Смакс) отбор образцов биологического материала в районе времени достижения максимальной концентрации действующего вещества (далее - Тмакс) проводится с частотой, достаточной для обеспечения статистической значимости исследования.
151. Продолжительность отбора проб биологического материала должна определяться по следующим критериям:
а) период полувыведения (продолжительность отбора проб биологического материала должна быть не менее чем в четыре раза больше периода полувыведения действующего вещества);
б) величина площади под фармакокинетической кривой (величина площади под фармакокинетической кривой (далее - ПФК) с момента введения лекарственного препарата до момента отбора последней пробы биологического материала для усредненной фармакокинетической кривой должна составлять не менее 80% от ПФК с момента введения лекарственного препарата до условной точки пересечения с осью времени).
152. До начала исследования биоэквивалентности лекарственных препаратов, действующие вещества которых являются эндогенными веществами, необходимо отобрать пробы биологического материала от каждого животного не менее двух раз в разные периоды времени с целью определения фонового уровня эндогенного вещества в организме животного.
153. В случае если для эндогенного вещества характерны суточные колебания концентрации в организме животного, необходимо определять концентрацию эндогенного вещества с частотой, достаточной для обеспечения статистической значимости исследования.
154. При исследовании биоэквивалентности лекарственного препарата, содержащего в составе несколько действующих веществ, следует определять концентрацию каждого действующего вещества, входящего в состав лекарственного препарата.
155. В случае если действующее вещество лекарственного препарата не обладает фармакологической активностью и эффективность лекарственного препарата определяется его метаболитом (метаболитами), следует определять концентрацию указанного метаболита (указанных метаболитов).
В случае если действующее вещество лекарственного препарата и его метаболит (метаболиты) обладают фармакологической активностью, следует определять концентрацию всех аналитов.
156. В случае если действующее вещество лекарственного препарата состоит из двух и более изомеров (или подвергается изомеризации в организме животного), которые обладают разной фармакокинетикой и фармакодинамикой, следует определять концентрацию каждого изомера. В случае если только один из изомеров обладает фармакологической активностью, при этом фармакологическая активность второго изомера низкая или полностью отсутствует, следует определять концентрацию только активного изомера.
157. Метод определения аналита в биологическом материале должен позволять определять концентрации аналита, соответствующие фармакологически активным концентрациям референтного лекарственного препарата, и быть линейным в указанном диапазоне концентраций.
158. Метод определения аналита должен быть валидирован.
159. Нижний предел количественного определения метода должен обеспечивать определение концентрации, не превышающей 5% от Смакс.
160. Для оценки сравнительной биодоступности исследуемых воспроизведенного и референтного лекарственных препаратов при определении скорости и степени абсорбции в исследованиях биоэквивалентности используется кривая "концентрация - время" и следующие фармакокинетические параметры с учетом их допустимых отклонений: ПФК, Смакс в биологическом материале, отношение Смакс к ПФК.
161. Референтный и воспроизведенный лекарственные препараты считаются биоэквивалентными, если отношение верхней и нижней границы доверительного интервала Смакс, ПФК и Смакс/ПФК воспроизведенного и референтного лекарственных препаратов находится в пределах 80 - 125% для Смакс и ПФК и 75 - 133% для Смакс/ПФК.