МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
СИНДРОМ ДЕФИЦИТА GLUT1
Кодирование по Международной статистической классификации болезней и проблем, связанных со здоровьем: G40.4
Год утверждения (частота пересмотра): 2024
Возрастная категория: Взрослые, Дети
Пересмотр не позднее: 2026
ID: 793
Разработчик клинической рекомендации
- Ассоциация медицинских генетиков
- Союз педиатров России
- Российская организация диетологов, нутрициологов и специалистов пищевой индустрии
Одобрено Научно-практическим Советом Минздрава РФ
Список сокращений
ГЭБ - гематоэнцефалический барьер
ДсНГИ - диета с низким гликемическим индексом
ЖКТ - желудочно-кишечный тракт
ИВД - диагностика in vitro
КД - кетогенная диета
КТ - компьютерная томография
МДА - модифицированная диета Аткинса
МРТ - магнитно-резонансная томография
ПЭП - противоэпилептический препарат
СМЖ - спинномозговая жидкость
УЗИ - ультразвуковое исследование
ЦНС - центральная нервная система
ЭЭГ - электроэнцефалография
BE (base excess) - избыток буферных оснований
CNS (Columbia Neurological Score) - неврологическая шкала Колумбийского университета
FDG-PET - ПЭТ с флудезоксиглюкозой [18F]
Glut1 (glucose transporter type 1) - однонаправленный белок-переносчик глюкозы
HGMD (Human Gene Mutation Database) - база данных мутаций в генах человека
LCT (long-chain triglycerides) - длинноцепочечные триглицериды
MCT (medium-chain triglycerides) - среднецепочечные триглицериды
MCT1 (monocarboxylate transporter 1) - переносчик монокарбоксилата 1
MLPA (multiplex ligation-dependent probe amplification - мультиплексная амплификация лигированных проб
RDA (recommended dietary allowances) - рекомендуемые нормы питания
Термины и определения
Кетогенная диета (КД) - метод диетотерапии, основанный на использовании рациона питания с высоким содержанием жиров и резко сниженным количеством углеводов, на фоне которого в организме активизируется выработка кетоновых тел.
Glut1 (glucose transporter type 1) - белок-переносчик глюкозы, отвечающий за ее транспорт из крови в головной мозг через гематоэнцефалический барьер.
1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
1.1 Определение заболевания или состояния (группы заболеваний или состояний)
Синдром дефицита Glut1 (синонимы: недостаточность Glut1, GLUT1 DS, болезнь де Виво) - наследственное заболевание, характеризующееся развитием ранней детской энцефалопатии, фармакорезистентной эпилепсии, формированием микроцефалии, задержкой психомоторного развития, атаксией, дизартрией, пароксизмальными дискинезиями (хореоатетоз/дистония) и альтернирующей гемиплегией различной степени выраженности.
1.2 Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
Причиной синдрома дефицита Glut1 являются мутации в гене SLC2A1, который локализован на коротком плече хромосомы 1 - 1p34.2. Ген SLC2A1, кодирующий белок Glut1, состоит из 10 экзонов и 9 интронов. В международной базе данных по мутациям человека HGMD (Human Gene Mutation Database - база данных мутаций в генах человека) описано более 300 различных мутаций в гене SLC2A1 [1]. Патогенные варианты представлены миссенс-мутациями, нонсенс-мутациями, небольшими внутригенными делециями/инсерциями, а также вариантами сайтов сплайсинга.
Ген SLC2A1 кодирует однонаправленный белок-переносчик глюкозы (Glut1), отвечающий за ее транспорт из крови в головной мозг через гематоэнцефалический барьер (ГЭБ).
В большинстве случаев причиной заболевания является гетерозиготная мутация в гене SLC2A1, возникшая de novo. Реже встречается передача патогенного варианта от родителя с легкой формой заболевания, обусловленной, вероятно, тканевым мозаицизмом [2]. Также описаны редкие случаи аутосомно-рецессивного типа наследования заболевания [3, 4].
Мутации в гене SLC2A1 влияют на функцию белка Glut1, что приводит к нарушению транспорта глюкозы в головной мозг, в результате чего он лишается основного источника энергии [5, 6].
Семейство белков Glut состоит из трех подклассов и включает 12 разновидностей белков. Данные белки-транспортеры облегчают пассивную диффузию глюкозы через тканевые барьеры путем энергозависимых механизмов. Glut1 экспрессируется в эндотелиальных клетках сосудов, входящих в состав ГЭБ, и отвечает за проникновение глюкозы в головной мозг. Glut2 ассоциирован с синдромом Фанкони-Биккеля. Glut3 отвечает за проникновение глюкозы через мембрану нейрональной плазмы, Glut4 является инсулин-регулирующим транспортером глюкозы жировой ткани, сердечной мышцы и скелетных мышц, отвечает за инсулин опосредованный транспорт глюкозы, Glut5 экспрессируется в кишечнике, тестикулах и почках. Функция Glut7 на данный момент неизвестна.
Белок Glut1 является ключевым посредником, ответственным за доставку глюкозы через ГЭБ [2]. На рисунке 1 представлены транспортеры для различных веществ через гематоэнцефалический барьер.
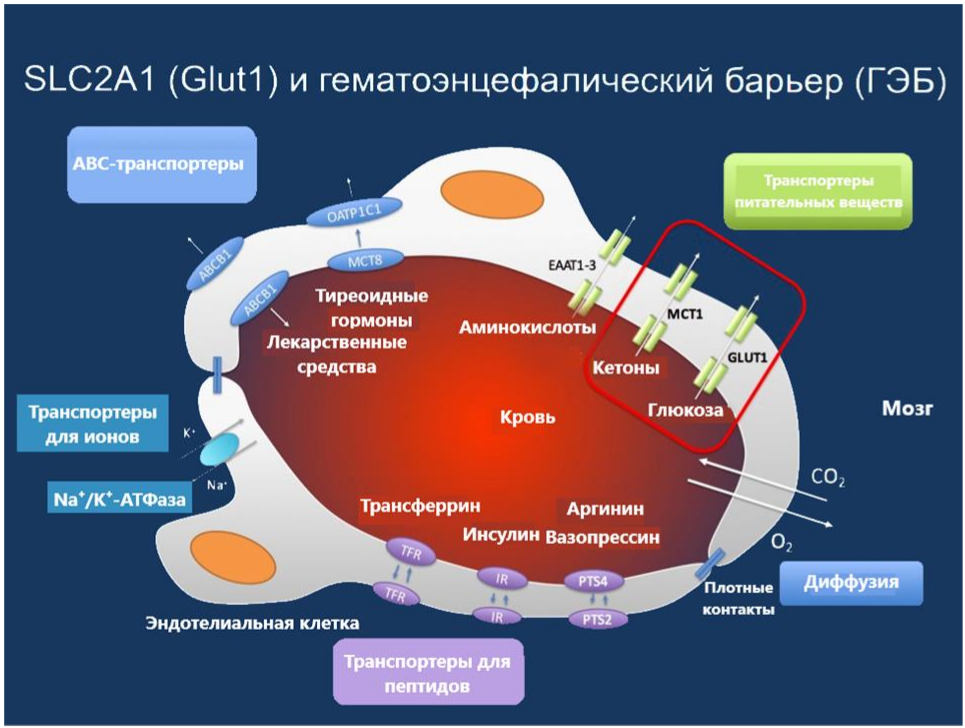
Рисунок 1 Транспорт различных веществ через ГЭБ
1.3 Эпидемиология заболевания или состояния (группы заболеваний или состояний)
Синдром дефицита Glut1 - это панэтническое заболевание, его частота по различным исследованиям составляет от 1:83 000 до 1:100 000 новорожденных [7, 8].
Исследования показали, что синдром дефицита Glut1 является причиной 10 - 12% случаев эпилепсии с абсансами (типичными и атипичными), 0,7 - 1% - тонико-клонических приступов, 0 - 5% - миоклонических, атонических и миоклонико-атонических приступов, 0,6% случаев развития приступов с рефрактерным течением и 2,7% случаев эпилепсий с интеллектуальным дефицитом и/или различными двигательными расстройствами [8, 9, 10, 11].
1.4 Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
Согласно МКБ-10, заболевание относится к VI классу - болезням нервной системы, эпизодическим и пароксизмальным расстройствам, другим видам генерализованной эпилепсии и эпилептических синдромов - G40.4.
Пароксизмальная дискинезия и эпилепсия, вызванные физической нагрузкой, ранее известные как дистония 18 - DYT18, или DYT-SLC2A1 [12], и пароксизмальный хореоатетоз со спастичностью - DYT9 [13], теперь признаны частью фенотипического спектра синдрома дефицита Glut1.
1.5 Классификация заболевания или состояния (группы заболеваний или состояний)
Варианты течения синдрома дефицита Glut1, различающиеся возрастом дебюта и неврологическими появлениями.
1. Классическая форма синдрома дефицита Glut1 (90% случаев)
Судороги возникают в возрасте от 1 месяца до 2 лет, реже - старше 2 лет. Данная форма характеризуется задержкой психомоторного развития, пароксизмальными движениями глаз (саккадирующими движениями и опсоклонусом глазных яблок), дизартрией, формированием микроцефалии и двигательными нарушениями, включая атаксию, альтернирующую гемиплегию, гиперкинезы по типу дистонии и хореи [14, 15];
2. Неклассическая форма синдрома дефицита Glut1 (10% случаев)
Пациенты имеют более мягкие проявления, чем при классической форме. Эпилептические приступы встречаются реже, более выражены пароксизмальные дискинезии: перемежающаяся атаксия, хореоатетоз, дистония и альтернирующая гемиплегия. В настоящее время известно несколько заболеваний, вызванных дефицитом Glut1 [15, 16, 17]:
1. Пароксизмальный хореоатетоз с эпизодической атаксией и спастичностью (наследственная дистония 9 типа - DYT9);
2. Пароксизмальная дискинезия, вызванная физическими упражнениями, с эпилепсией или без нее (наследственная дистония 18 типа - DYT18; синдром дефицита Glut1 2 типа);
3. Фенотип атипичной абсансной эпилепсии детского возраста;
4. Фенотип эпилепсии с миоклонико-атоническими приступами.
Некоторые клинические проявления данных заболеваний могут совпадать с наблюдаемыми при классическом синдроме дефицита Glut1 [18].
1.6 Клиническая картина заболевания или состояния (группы заболеваний или состояний)
Для классического варианта синдрома дефицита Glut1 характерна манифестация в первые месяцы жизни. Эпилептические приступы полиморфные: тонико-клонические, миоклонические, атипичные абсансы, атонические и миоклонико-атонические, могут возникать с различной частотой - от ежемесячных до ежедневных, характеризуются выраженной резистентностью к терапии противоэпилептическими препаратами. Возможны эпизоды апноэ, цианоза, пароксизмальные движения глаз. В последующем присоединяются двигательные нарушения (атаксия, дистония, нарушения мышечного тонуса по спастическому типу), формируется микроцефалия. На электроэнцефалограмме (ЭЭГ) нередко выявляются генерализованные или фокальные эпилептиформные изменения. Важным патогномоничным признаком заболевания является снижение частоты эпилептических приступов и изменений при ЭЭГ после приема пищи [19].
Эпилепсия при синдроме дефицита Glut1 достигает своего пика в первые годы жизни, затем по мере роста ребенка судороги становятся менее частыми или даже исчезают у некоторых пациентов к подростковому и взрослому возрастам. Характер приступов со временем меняется: фокальные приступы чаще встречаются в младенчестве, атипичные абсансы и миоклонические приступы начинаются со второго года жизни, а генерализованные тонико-клонические приступы обычно появляются после 3-х лет [2]. Задержка развития, атаксия и микроцефалия возникают в младенчестве или раннем детстве. Дистония - единственный симптом, частота которого постоянно нарастает, проявляясь в позднем детстве или подростковом возрасте [14]. Пароксизмальные неврологические явления, как правило, начинаются в раннем детстве, по мере взросления пациентов становятся легче и реже, а в некоторых случаях они могут совсем исчезнуть [20, 21, 12, 22, 23]. У 18% пациентов с синдромом дефицита Glut1 описаны обсессивно-компульсивные расстройства [2].
При неэпилептическом течении заболевания доминируют двигательные расстройства: пароксизмальные дискинезии (хореоатетоз/дистония/тремор), атаксия, альтернирующие гемиплегии различной степени выраженности. В зависимости от сроков манифестации частота различных симптомов может варьировать (приложение Г1).
В настоящее время синдром дефицита Glut1 рассматривается как заболевание с континуумом клинических фенотипов, различающихся по возрасту манифестации, тяжести клинических проявлений и скорости прогрессирования [24, 25]. Генотип-фенотипическая корреляция при синдроме дефицита Glut1 представлена в приложении Г2.
Эпилепсия при синдроме дефицита Glut1
Фармакорезистентная форма эпилепсии часто является первым симптомом синдрома дефицита Glut1 [2, 26]. При этом могут наблюдаться любые типы эпилептических приступов [27]. Наиболее часто наблюдаются генерализованные эпилептические приступы, реже фокальные [28, 29]. Ранняя манифестация абсансных приступов (до 4-х лет) и эпилепсия с миоклонико-атоническими приступами (синдром Дузе) нередко обусловлены патогенными мутациями гена SLC2A1. Эпилептические приступы при классическом синдроме дефицита Glut1 дебютируют в возрасте от одного до шести месяцев и часто являются первым клиническим симптомом. У некоторых детей эпилептическим приступам предшествуют эпизоды апноэ и пароксизмальные движения глазных яблок, похожие на опсоклонус [30]. В младенчестве фокальные приступы проявляются как пароксизмальными движениями головы и глазных яблок, а также по типу сложных атипичных абсансов и атонических приступов. ЭЭГ может быть представлена фокальными, мультифокальными и генерализованными разрядами.
По мере роста ребенка могут отмечаться несколько видов приступов: генерализованные и фокальные тонические, клонические, тонико-клонические, миоклонические, атипичные абсансы, атонические и неклассифицированные [31]. В ЭЭГ может регистрироваться региональная, мультирегиональная, генерализованная активность (в том числе и генерализованная ритмичная активность частотой 2 - 4 Гц, соответствующая атипичным и типичным абсансам).
Частота приступов варьирует среди пациентов: у некоторых они наблюдаются ежедневно, у других отмечаются единичные приступы с интервалом в дни, недели, месяцы или годы. Частота приступов не коррелирует с тяжестью заболевания. Приступы часто сочетаются с задержкой развития или атаксией.
Клинические фенотипы синдрома дефицита Glut1 часто имеют фенотипическое сходство с классическими синдромами идиопатической генерализованной эпилепсии, такими как детская абсансная эпилепсия, юношеская абсансная эпилепсия, юношеская миоклоническая эпилепсия и, реже, эпилепсия с изолированными судорожными приступами [32].
Тот факт, что мутации в гене SLC2A1 чаще встречаются у больных эпилепсией с началом в раннем детском возрасте, включая абсансную эпилепсию с ранним дебютом и миоклонико-атоническую эпилепсию, наводит на мысль о возможных механизмах возрастной зависимости [2, 24, 32, 33, 34, 35]. Это явление может быть объяснено созреванием головного мозга и транспорта глюкозы, что доказано с помощью позитронно-эмиссионной томографии [36], и возрастной экспрессией гена SLC2A1 [18]. Таким образом, можно констатировать, что молекулярно-генетический анализ гена SLC2A1 оправдан у всех детей с абсансными приступами, особенно в случае их начала до 4-летнего возраста. Это позволит начать эффективное лечение раньше и провести генетическое консультирование семьи [24].
Интеллектуальные нарушения
У пациентов с синдромом дефицита Glut1 наблюдаются когнитивные нарушения различной степени тяжести, начиная от трудностей в обучении и заканчивая тяжелым интеллектуальным дефицитом. Фармакорезистентная эпилептическая энцефалопатия при синдроме дефицита Glut1 обычно приводит к нарушениям интеллекта, изолированный интеллектуальный дефицит наблюдается редко.
Речевые и языковые нарушения развития различной степени выраженности наблюдаются у всех пациентов. Распространена дизартрия, сопровождающаяся дисфлюэнтом (чрезмерно прерванной речью). Нарушены перцептивные и экспрессивные языковые навыки.
Социально-адаптивное поведение не страдает, пациенты обычно чувствуют себя комфортно в группе и в условиях школы и хорошо общаются с другими. Расстройства аутистического спектра наблюдаются редко.
Двигательные нарушения
Двигательные расстройства характеризуются атаксией, дистонией и хореей, могут быть постоянными или пароксизмальными [16, 37, 38, 39]. Важным диагностическим критерием является их тенденция к ухудшению во время голода, физических нагрузок или при воздействии других факторов окружающей среды, таких как интеркуррентные заболевания, инфекции, лихорадка, беспокойство и нарушение диеты. Иногда провоцирующий триггер невозможно обнаружить [37, 38, 40, 41, 42, 43, 44].
Варианты двигательных нарушений при синдроме дефицита Glut1:
- Атаксия
- Атаксия с пирамидными симптомами
- Дистония
- Хорея
- Тремор
- Миоклонус
- Диспраксия
- Тики
- Стереотипии
- Пароксизмальные двигательные расстройства
- Пароксизмальные явления с выраженной двигательной дисфункцией
- Пароксизмальные дискинезии, вызванные физической нагрузкой
- Пароксизмальные движения глаз
- Пароксизмальные явления со сложными неврологическими симптомами
Другие пароксизмальные явления:
- Мигрень
- Циклическая рвота
- Депривация сна [45].
Нарушения походки
У пациентов с синдромом дефицита Glut1, как правило, отмечается задержка моторного развития. Они начинают позже ходить, а пациенты с тяжелым вариантом заболевания не способны ходить самостоятельно [9, 20, 30, 40, 42, 44, 46, 47]. Типичным нарушением при походке при синдроме дефицита Glut1 является атаксия [2, 6, 23, 48, 49]. Одновременно наблюдается спастичность и, как следствие, спастически-атаксическая походка, которая характеризуется неустойчивостью, скованностью ног, укороченной длиной шага и уменьшенным ударом пятки, а также походка на широкой опоре (в отличие от походки "ножницы" при спастической диплегии при церебральном параличе) [48, 50]. У части пациентов наблюдается дистоническая поза верхних конечностей при ходьбе, у других - хореоатетоз [48]. Другие, менее распространенные нарушения походки, наблюдаемые при тяжелых фенотипах синдрома дефицита Glut1, включают спастическую и, преимущественно, дистоническую походку [6, 48, 51]. Характерно эпизодическое нарушение походки при физической нагрузке или голодании. Некоторые авторы описывают неспособность долго стоять из-за атаксии, которая уменьшается после приема пищи [24, 48]. Это периодическое нарушение походки напоминает другие генетические нарушения, такие как болезнь Сегавы, и наводит на мысль о возможном общем патофизиологическом механизме заболевания, например, синаптической дисфункции. Синдром дефицита Glut1 не считается нейродегенеративным заболеванием, но может наблюдаться прогрессирование спастичности и, реже, атаксии в период полового созревания. Это ухудшение может быть временным [2, 14, 22, 52].
Дистония
Дистония - второе по частоте двигательное расстройство при синдроме дефицита Glut1. Сообщается, что дистония встречаются у 20 - 86% пациентов с классическим фенотипом и у 13% пациентов с неклассическим фенотипом [2, 6, 23, 33, 48, 53]. Степень тяжести дистонии при синдроме дефицита Glut1 варьирует, и, в основном, представляет собой постуральную дистонию или дистонию действия, затрагивающую дистальные конечности, чаще верхние. У некоторых пациентов сообщалось о специфической дистонии и дистоническом треморе [2, 12, 17, 25, 51, 52, 54, 55].
Тремор
Тремор встречается более, чем у 70% пациентов с синдромом дефицита Glut1 с классическим фенотипом [48]. Он характеризуется как тремор терминального действия и сочетается с атаксией, дизартрией, диссинергией, нарушением координации движений туловища и глазной диспраксией, что отражает выраженность дисфункции мозжечка при этом заболевании [48, 56, 57]. У части пациентов наблюдались постуральный и дистонический тремор. Признаки паркинсонизма являются редким симптомом при синдроме дефицита Glut1, о случаях с тремором в состоянии покоя не сообщалось [20, 35, 51, 55].
Хореические гиперкинезы описаны у 3 - 75% пациентов по данным разных публикаций, что, вероятно, отражает предвзятость установления данного симптома [2, 6, 48]. Хорея часто бывает легкой, затрагивает только лицо и дистальные отделы верхних конечностей.
Миоклонус
Миоклонус при синдроме дефицита Glut1 обычно имеет эпилептическое происхождение. Однако неэпилептический миоклонус, включая испуг, движение и постуральный миоклонус, также был зарегистрирован у отдельных пациентов [32, 48, 49].
Диспраксия
У пациентов с синдромом дефицита Glut1 часто наблюдается диспраксия, характеризующаяся трудностями двигательного планирования при выполнении конкретных движений. Глазная и орально-буккальная диспраксия описана у 20% пациентов с классическим фенотипом [48], характеризуется нарушением координации глаз и рук, дезорганизацией мелкой моторики и неуклюжестью [2, 25].
Стереотипии и тики
Стереотипии и тики были описаны у некоторых пациентов с синдромом дефицита Glut1 [48], но не являются специфическими симптомами заболевания, учитывая высокую распространенность этих доброкачественных двигательных нарушений в общей популяции.
Пароксизмальные двигательные расстройства
Пароксизмальные неэпилептические перемежающиеся неврологические симптомы, часто с выраженными моторными проявлениями, описаны у пациентов с синдромом дефицита Glut1 с разными фенотипами. Частота пароксизмальных неэпилептических приступов составляет от 30% до 59% случаев [6, 23, 49]. Несмотря на то, что такие пароксизмальные состояния возникают у пациентов с разными фенотипами, принято считать, что они более характерны для мягких форм синдрома дефицита Glut1 [17, 21].
Недостаточное поступление энергии, приводящее, вероятно, к синаптической дисфункции, является предполагаемым механизмом, лежащим в основе пароксизмальных приступов при синдроме дефицита Glut1. В связи с чем провоцирующие факторы включают в себя состояния, требующие повышенных энергозатрат, такие как состояние после пробуждения или перед едой, голодание, повышенная физическая активность. Другими частыми провоцирующими факторами являются эмоциональный стресс, лихорадка, усталость или несоблюдение кетогенной диеты. Сообщалось также о депривации сна, изменениях температуры и факторах, связанных с приемом лекарств (например, фенобарбитала**, клоназепама**, теофиллина). Иногда триггеры не выявляются. Наиболее частыми смягчающими факторами являются прием пищи, потребление углеводов и отдых [2, 6, 12, 17, 21, 23, 32, 48, 49, 51].
Клинические проявления пароксизмальных приступов, связанных с синдромом дефицита Glut1, очень разнообразны. Однако у каждого пациента они, как правило, стереотипны. Нозологически они не имеют эпилептической природы. Некоторые пациенты могут испытывать выраженную дисфорию и эмоциональную лабильность во время приступов, в то время как другие испытывают замешательство или сонливость. Эпизоды могут представлять собой тип фокального приступа, который не обнаруживается с помощью ЭЭГ, но факторы, опровергающие эту теорию, включают сохранное сознание, отсутствие других типичных клинических проявлений приступов и нормальную иктальную ЭЭГ [48]. Также могут возникать немоторные пароксизмальные явления, такие как мигрень, эпизоды поведенческих и эмоциональных расстройств, циклические рвоты или эпизоды внезапно наступающего сна [2, 22, 23, 25, 48, 49].
Эпизоды могут начинаться постепенно или молниеносно. При постепенном прогрессировании эпизоды проявляются заметным ухудшением исходного неврологического статуса. В целом, пациенты с классическим течением заболевания имеют тенденцию к развитию несколько типов пароксизмальных приступов, тогда как пациенты с неклассическим течением склонны проявлять один-два типа приступов. Продолжительность эпизодов колеблется от нескольких минут до часов, а иногда и дней, причем события, возникающие молниеносно, короче по продолжительности. Частота варьирует от ежедневного до еженедельного, ежемесячного или раз в несколько месяцев [2, 17, 20, 21, 23, 25, 48, 49, 51, 52, 58].
При классическом фенотипе пароксизмальные двигательные расстройства, как правило, развиваются позже в детстве или в подростковом возрасте, когда эпилептические приступы становятся менее выраженными или исчезают. При неклассических фенотипах эпизоды обычно начинаются в раннем или позднем детском возрасте. В целом у пациентов наблюдается постепенное улучшение по мере взросления, а в некоторых случаях явления могут полностью исчезать, независимо от того, придерживается ли пациент кетогенной диеты [2, 12, 14, 17, 20 - 23, 25, 51, 55].
Пароксизмальные явления с серьезной двигательной дисфункцией
В эту группу пароксизмальных состояний входят периодические эпизоды слабости, атаксии и некинезигенных дискинезий. Периодические эпизоды слабости, проявляющиеся в виде парапареза, тетрапареза, гемипареза или монопареза, наблюдаются у 29 - 50% пациентов с синдромом дефицита Glut1 [2, 49, 59]. Также может возникнуть внезапный общий паралич тела, имитирующий периодический паралич [17, 18, 23, 48].
Патофизиология, лежащая в основе преходящих неврологических симптомов, не определена. При нейровизуализации регистрируют признаки очаговой гипоперфузии головного мозга [59]. Были предложены два механизма этого явления: гипометаболизм, связанный с недостаточным поступлением глюкозы в центральную нервную систему, дисфункция церебральных кровеносных сосудов и предрасположенность к преходящему спазму сосудов. Последнее подтверждается известным обилием переносчиков Glut1 в церебральных эндотелиальных клетках, нарушением церебрального ангиогенеза и результирующим уменьшением микрососудов головного мозга на мышиной модели с гаплонедостаточностью SLC1A2 [37].
Иногда картина эпизодов гемиплегии и сопутствующие клинические проявления у пациентов с синдромом дефицита Glut1 могут имитировать синдром альтернирующей гемиплегии в детском возрасте [2, 60, 61]. Однако, в отличие от классической альтернирующей гемиплегии в детстве, возраст проявления атипичных приступов, связанных с синдромом дефицита Glut1, обычно старше, во время приступов не наблюдается выраженной вегетативной или бульбарной дисфункции, а эпизоды часто возникают из-за голодания или физических упражнений и смягчаются после приема пищи, особенно богатой углеводами. Эпизоды пароксизмальной некинезигенной дискинезии при синдроме дефицита Glut1 проявляются либо хореей, либо дистонией, часто затрагивающей конечности, а иногда и аксиальную или орофациальную мускулатуру [22, 23, 25, 48, 49]. Также могут возникать приступы пароксизмальной атаксии, которые в некоторых случаях могут напоминать эпизодическую атаксию [6, 20, 32, 36, 62].
Пароксизмальные дискинезии, вызванные физической нагрузкой
Дискинезия и эпилепсия, вызванные пароксизмальной физической нагрузкой, ранее известная как дистония 18 - DYT18 [20, 21, 48, 49], и пароксизмальный хореоатетоз со спастичностью, ранее известный как дистония 9 [12], в настоящее время признаны частью фенотипического спектра синдрома дефицита Glut1.
Пароксизмальная дискинезия и эпилепсия, вызванные физической нагрузкой, клинически отличаются от классического синдрома дефицита Glut1 тем, что у большинства пораженных лиц нормальное межприступное неврологическое состояние и нормальная окружность головы, а дискинезия, вызванная физической нагрузкой, развивается у них в более позднем возрасте [20, 21, 48, 49]. Концентрации глюкозы в спинномозговой жидкости (СМЖ), как правило, выше (41 - 52 мг/дл), чем при классическом синдроме дефицита Glut1 [12].
Описаны две семьи с пароксизмальным хореоатетозом со спастичностью, которые имели пароксизмальную, главным образом, вызванную физической нагрузкой, дискинезию в возрасте от одного года до 15 лет [22], обусловленную гетерозиготными патогенными вариантами в гене SLC2A1 - p.Arg212Cys и p.Arg126Cys. Триггерами дискинезии являлись длительные физические нагрузки, чувство тревоги и эмоциональный стресс. С возрастом дискинезии возникали реже или прекращались. Среди сопутствующих симптомов отмечались прогрессирующий спастический парапарез, легкая атаксия, когнитивные нарушения легкой и средней степени тяжести и эпилептические приступы.
В литературе описаны и другие приступообразные явления [21, 23, 37, 47]. Однако неясно, представляют ли эти события эпилептические или неэпилептические явления. Неврологические признаки, которые обычно варьируют и могут зависеть от таких факторов, как голод или усталость, включают следующие:
- спутанность сознания;
- вялость;
- сонливость;
- периодические головные боли, мигрени;
- нарушения сна;
- односторонний парез;
- тотальный паралич тела;
- перемежающаяся атаксия;
- хорея;
- дистония действия конечностей;
- мозжечковый акционный тремор;
- писчий спазм;
- дистонический тремор (описывается как единственный признак у матери и дочери) [63];
- паркинсонизм;
- миоклонус;
- диспраксия;
- некинезигенные дискинезии.
Пароксизмальные движения глазных яблок
У пациентов с синдромом дефицита Glut1 отмечаются пароксизмальные аномальные движения глазных яблок [25, 30, 31, 51, 80]. Аномалии движения глаз являются первым неврологическим признаком у 38% пациентов [32]. На основе ретроспективного анализа 101 пациента с генетически подтвержденным синдромом дефицита Glut1 и видеоанализом эпизодов движения глаз описаны характеристики пароксизмальных аномальных движений глаз в младенчестве [30].
Движения глаз быстрые, разнонаправленные и часто сопровождаются движениями головы в одном направлении. Движения всегда четко разделены во времени интервалами, обычно в диапазоне от 200 до 800 мс, что соответствует средней частоте движения глаз - примерно 2 в секунду. Эти особенности наиболее соответствуют саккадическим сдвигам взгляд-голова, которые характеризуются наличием межсаккадических интервалов, совмещенным направлением движения глаза и головы и необязательным присутствием компонента головы. Пароксизмальные саккады взгляда глаз-голова могут быть специфическим признаком синдрома дефицита Glut1 в младенчестве.
Движение глазных яблок при синдроме дефицита Glut1 было описано как опсоклонус. Однако в отличие от наблюдаемых авторами движений глаз, опсоклонус не имеет интервала фиксации между движениями и не связан с движением головы в одном направлении. Патофизиологический механизм, лежащий в основе пароксизмальных саккад при синдроме дефицита Glut1, неизвестен. Смещение взгляда обычно служит для того, чтобы подвести интересующий объект, обнаруженный в периферическом поле зрения, к фовеа, где его можно разглядеть более подробно. В зрелой нервной системе управление взглядом включает как активные сигналы, управляющие глазами, так и активные сигналы, подавляющие движения глаз и, следовательно, способствующие фиксации [34]. Сдвиг взгляда часто включает как саккады глаз, так и движения головы. Сигналы для движений головы и глаз берут начало в ретикулярной формации парамедианного моста (для горизонтальных движений глаз) и в мезэнцефалической ретикулярной формации (для вертикальных движений глаз) [35]. Нейронная сеть управления взглядом включает верхний холмик четверохолмия, лобную долю и заднюю теменную кору [16, 18, 36, 37]. Дорсальное ядро шва подавляет генераторы саккад ствола мозга, ретикулярная часть черной субстанции подавляет верхний бугорок, а нейроны в лобной доле определяют объекты в поле зрения, которые являются неподходящими целями для саккад [38, 39, 48].
Вышеупомянутые эпизоды характеризуются явными непроизвольными повторяющимися саккадами взгляда. Симптомы почти всегда проявляются в течение первых 6 месяцев жизни, когда зрительная система быстро созревает и развивается способность подавлять рефлексивные саккады на зрительные стимулы [49]. Поскольку нейроны дорсального шва и части черной субстанции проводят импульсы с высокой скоростью, за исключением движений глаз, возможно, что их активность вызвана дефицитом Glut1, что делает возможным возникновение несоответствующих саккад. Нехватка энергии - один из механизмов, который, как предполагается, лежит в основе других пароксизмальных событий при синдроме дефицита Glut1 [14]. В соответствии с этой гипотезой, эпизодические движения глаз были вызваны усталостью, возбуждением или голоданием и у некоторых пациентов исчезали на фоне кетогенной (КД).
Вероятность того, что эти эпизоды представляют собой тип фокального припадка, который не обнаруживается с помощью накожной ЭЭГ, не может быть полностью исключена, но маловероятна. Отсутствие других типичных клинических проявлений приступов и нормальная иктальная ЭЭГ позволяют предположить, что эти явления неэпилептические. У большинства пациентов была сопутствующая эпилепсия, которая проявлялась в том же возрасте, что и эпизоды движения глазных яблок у 10 пациентов. У 6 пациентов первый приступ произошел только через 7 - 21 месяц после начала эпизодов движения глаз.
Эпизоды движения глазных яблок возникли в возрасте до 6 месяцев у 83% исследуемых пациентов. Среди 8 пациентов, у которых было известно течение эпизодов, частота эпизодов снизилась к позднему младенчеству и исчезла у всех, кроме одного, к 8 годам. Таким образом, эти эпизоды представляют собой зависящее от возраста проявление заболевания, которое, вероятно, связано с определенной стадией развития мозга.
Краткие пароксизмальные эпизоды движений глаз и головы, для которых мы предлагаем термин аберрантные саккады взгляда, являются характерной и ранней особенностью синдрома дефицита Glut1 в младенчестве. Неспособность удовлетворить потребность в энергии является вероятным патофизиологическим механизмом. Поскольку ранняя диагностика улучшает долгосрочный прогноз пациентов с синдромом дефицита Glut1 жизненно важно, чтобы неврологи распознавали эти эпизоды как ранний диагностический критерий заболевания [65].
1.7 Дифференциальный диагноз
Дифференциальный диагноз синдрома дефицита Glut1 включает в себя следующие состояния:
- другие причины снижения концентрации глюкозы в СМЖ, включая состояния, вызывающие хроническую или транзиторную гипогликемию (например, семейный гиперинсулинизм);
- все состояния с судорогами новорожденных и приобретенной микроцефалией; в частности, ранние формы синдрома Ретта, синдром Ангельмана и инфантильные формы нейронального цероидного липофусциноза;
- синдром опсоклонус-миоклонус [38, 57];
- неуточненные эпилептические энцефалопатии с задержками развития;
- семейные эпилепсии с аутосомно-доминантным типом наследования;
- эпизоды пароксизмальной неврологической дисфункции, реагирующей или предотвращаемой приемом углеводов, особенно при проявлениях в виде чередующегося гемипареза, атаксии, когнитивной дисфункции или судорог;
- другие расстройства движений, включая дистонию.
2. Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
Диагноз синдром дефицита Glut1 устанавливается на основании совокупности анамнестических данных, клинических данных, результатов лабораторных исследований (биохимических и молекулярно-генетических).
Обращаем внимание, что, согласно требованиям к разработке клинических рекомендаций, к каждому тезису-рекомендации необходимо указывать силу рекомендаций и доказательную базу в соответствии со шкалами оценки уровня достоверности доказательств (УДД) и уровня убедительности рекомендаций (УУР). Для многих тезисов УУР и УДД будет низким по причине отсутствия посвященных им клинических исследований высокого дизайна. Невзирая на это, они являются необходимыми элементами обследования пациента для установления диагноза и выбора тактики лечения.
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб важно обратить внимание на следующие жалобы и анамнестические события:
- Отягощенный семейный анамнез (сходные симптомы у одного из родителей пробанда, проявляющиеся в легкой степени)
- Задержка психомоторного развития с медленным приобретением психомоторных навыков)
- Судороги
- Нарушение речи
- Нарушения движений в конечностях
- Нарушение походки
Жалобы при классическом GLUT1 (~ 90%)
- Приступы с началом в возрасте до двух лет (чаще всего в возрасте от 1 до 6 месяцев)
- Задержка психомоторного развития
- Пароксизмальные движения глазных яблок (опсоклонус)
- Дизартрия
- Приобретенная микроцефалия
- Сложные двигательные расстройства, в том числе:
- Атаксия
- Дистония
- Хорея
- Тремор
Жалобы при неклассическом GLUT1 (~ 10%)
- Частые пароксизмальные дискинезии, в том числе:
- Пароксизмальная атаксия
- Хореоатетоз
- Дистония
- Альтернирующая гемиплегия
- Пароксизмальное головокружение
- Пароксизмальный паралич
- Жалобы, соответствующие клинике паркинсонизма
- Пароксизмальные боли в мышцах по ночам
- Эпизоды повторных рвот
- Инсультоподобные состояния
Пациенты также жалуются на головную боль. Значительно реже отмечается снижение остроты зрения [49, 66]. Типично усиление клинической симптоматики в периоды гипертермии, после физической нагрузки, при голодании и интеркуррентных заболеваниях. У всех пациентов с синдромом дефицита Glut1 наблюдается прогрессирующая задержка общего развития. Нарушается интеллект, вербальные функции (дизартрия) и развиваются изменения в двигательной сфере [18, 67].
2.2 Физикальное обследование
При осмотре необходимо обратить внимание на следующие основные клинические проявления синдрома дефицита Glut1:
- Микроцефалия (встречается у половины пациентов)
- Эпилептические приступы
- Дизартрия
- Атаксия
- Хореоатетоз
- Тремор
- Нарушения мышечного тонуса по спастическому типу
- Альтернирующая гемиплегия
- Мышечная гипотония
- Дистонии
- Пароксизмальные движения глазных яблок
- Головокружение
- Гепатоспленомегалия.
2.3 Лабораторные диагностические исследования
Основные лабораторные методы подтверждения диагноза включают биохимические исследования и молекулярно-генетические исследования (поиск мутаций в гене SLC2A1).
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 проводить исследование уровня глюкозы в спинномозговой жидкости с параллельным исследованием уровня глюкозы в крови с целью уточнения диагноза [2, 17, 22, 25, 50].
Уровень убедительности рекомендаций A (уровень достоверности доказательств - 2)
Комментарии: При синдроме дефицита Glut1 отмечается снижение уровня глюкозы в СМЖ ниже 60 мг/дл (диапазон: 16,2 - 52 мг/дл); в более чем 90% - ниже 40 мг/дл и приблизительно в 10% она составляет 41 - 52 мг/дл. Исследование проводится после четырехчасового голодания. Образец крови получают непосредственно перед проведением люмбальной пункции: концентрация глюкозы в крови должна быть нормальной для исключения гипогликемии как причины гипогликорахии. Патогномоничным для синдрома дефицита Glut1 (встречается в 90% случаев) считается содержание глюкозы в СМЖ < 2,2 ммоль/л.
Соотношение глюкозы в СМЖ / глюкозы в крови у пациентов обычно менее 0,4 (в диапазоне 0,19 - 0,59); однако это значение менее информативно, чем абсолютное значение глюкозы СМЖ.
Уровни глюкозы в СМЖ 41 - 52 мг/дл могут наблюдаться при более мягких фенотипах [24, 62]. Значения глюкозы в СМЖ менее 60 мг/дл (3,3 ммоль/л) следует рассматривать как вероятные отклонения от нормы.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 определение вариантов генов в образце биологического материала другом или неуточненном неклассифицированных в других рубриках методом секвенирования по Сенгеру (03.Я99.18.999.039) с целью подтверждения диагноза на молекулярно-генетическом уровне с целью подтверждения генетической причины заболевания [68, 88, 92].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: диагноз синдром дефицита Glut1 выставляется на основании клинической картины, нормативных значений уровня глюкозы в крови, сниженного содержания глюкозы в СМЖ (< 60 мг/дл) и при выявлении патогенного варианта в гене SLC2A1 в гетерозиготном состоянии. Заболевание наследуется по аутосомно-доминантному типу. 84% пациентов имеют точечные мутации в гене SLC2A1, которые можно выявить преимущественно методом массового параллельного секвенирования (анализ экзома или таргетные панели генов) или методом прямого секвенирования по Сенгеру.
У 90% пациентов заболевание развивается вследствие спорадического (de novo) патогенного варианта в гене SLC2A1. В 10% случаев мутация унаследована от одного из родителей, бессимптомного или с умеренными проявлениями болезни, что является следствием тканевого мозаицизма.
В случае отсутствия генетического подтверждения диагноза, но при наличии характерной клинической картины синдрома дефицита Glut1, а также снижения уровня глюкозы в ликворе, показано придерживаться клинических рекомендаций по синдрому дефицита Glut1.
Риск возникновения заболевания для детей пробанда с синдромом дефицита Glut1 составляет 50%.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 определение вариантов генов в образце биологического материала другом или неуточненном, неклассифицированные в других рубриках, методом множественной лигазно-зависимой амплификации зондов (03.Я99.18.999.059) с целью подтверждения генетической причины заболевания [88, 92].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: в случае отрицательных результатов секвенирования по Сэнгеру проводится исследование методом MLPA.
У 13% пациентов выявляют крупные делеции или дупликации гена SLC2A1, для детекции которых и используют метод MLPA (multiplex ligation-dependent probe amplification - мультиплексная амплификация лигированных проб) [17].
В случае отсутствия генетического подтверждения диагноза, но при наличии характерной клинической картины синдрома дефицита Glut1, а также снижения уровня глюкозы в ликворе, показано придерживаться клинических рекомендаций по синдрому дефицита Glut1.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 проведение общего (клинического) анализа крови развернутого для оценки основных параметров кроветворения и наличия воспалительных процессов при подборе терапии [3, 69, 70, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: необходимо исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула) и исследование скорости оседания эритроцитов. Атипичные варианты течения в редких случаях могут сопровождаться гемолитической анемией. Исследование проводится в процессе динамического наблюдения, частота определяется индивидуально.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 проведение анализа крови биохимического общетерапевтического (исследование уровня глюкозы в крови) для своевременной диагностики гипогликемических состояний [69, 72, 77, 92]
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: необходимо провести исследование уровня общего белка в крови, исследование уровня альбумина в крови, исследование уровня мочевины в крови, исследование уровня креатинина в крови, исследование уровня триглицеридов в крови, исследование уровня холестерина в крови, исследование уровня холестерина липопротеинов низкой плотности, исследование уровня липопротеинов в крови, определение активности щелочной фосфатазы в крови, исследование уровня общего билирубина в крови, исследование уровня свободного и связанного билирубина в крови, исследование уровня 25-OH витамина Д в крови, исследование уровня глюкозы в крови, определение активности аспартатаминотрансферазы в крови, определение активности аланинаминотрансферазы в крови, определение активности амилазы в крови, исследование уровня натрия в крови, исследование уровня калия в крови, исследование уровня общего кальция в крови, исследование уровня общего магния в сыворотке крови. При синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать результаты анализа крови биохимического общетерапевтического.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 исследование кислотно-основного состояния и газов крови крови (исследование уровня водородных ионов (pH) крови, исследование уровня буферных веществ в крови), уровня глюкозы в крови, исследование уровня натрия в крови, уровня общего кальция в крови, уровня калия в крови, уровня хлоридов в крови, исследование уровня молочной кислоты в крови с целью дифференциальной диагностики состояния, диагностики метаболических нарушений перед введением в КД [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: На этапе введения кетогенной диеты данные параметры оцениваются 1 раз в 1 - 3 дня, далее при стабильном кетозе по показаниям.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 обнаружение кетоновых тел в моче или обнаружение кетоновых тел в моче экспресс-методом для оценки их исходного уровня перед введением в кетогенную диету [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: При синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать состояние внутренних органов.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 и подтвержденным синдромом дефицита Glut1 обнаружение кетоновых тел и исследования уровня глюкозы в крови с помощью системы мониторинга глюкозы/кетонов в крови ИВД (диагностика in vitro) для домашнего использования для оценки эффективности кетогенной диеты - определение исходного уровня перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: для оценки эффективности КД необходим регулярный самоконтроль уровня кетоновых тел и глюкозы в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня кетонов и глюкозы.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 и подтвержденным синдромом дефицита Glut1 исследование уровня глюкозы в крови с помощью системы мониторинга глюкозы ИВД для домашнего использования или системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования для оценки эффективности кетогенной диеты - перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: для оценки эффективности КД необходим регулярный самоконтроль уровня глюкозы в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня глюкозы.
2.4 Инструментальные диагностические исследования
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 проведение электроэнцефалографии при установлении диагноза с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: ЭЭГ должна быть проведена как минимум за 3 месяца до назначения КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 проведение электроэнцефалографии с видеомониторингом до еды, во время и после приема пищи с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности при невозможности проведения стандартной (рутинной) электроэнцефалографии [73].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3)
Комментарии: ЭЭГ с видеомониторингом должна быть проведена при невозможности проведения стандартной (рутинной) ЭЭГ как минимум за 3 месяца до начала КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
У пациентов с Glut1 интериктальная ЭЭГ обычно в норме. По результатам ЭЭГ чаще всего определяются мультифокальные спайк-волновые разряды, полиспайк-волновые разряды и билатерально синхронные.
У младенцев чаще регистрируется региональное замедление и эпилептиформные разряды. У детей в возрасте двух лет и старше наблюдается генерализованная спайк-волна с частотой 2,5 - 4 Гц [27, 31, 74].
Может наблюдаться любой тип приступов, чаще встречаются генерализованные тонические или клонические, абсансы, фокальные, миоклонические, атонические.
Важным патогномоничным признаком заболевания является регресс эпилептических приступов и патологических изменений по ЭЭГ после приема пищи.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 проведение магнитно-резонансной томографии головного мозга (МРТ) для определения степени тяжести заболевания и дифференциальной диагностики с нейродегенеративными и другими заболеваниями [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: магнитно-резонансная томография проводится с целью исключения структурных аномалий головного мозга и нейрометаболических заболеваний. При нейровизуализации у пациентов с синдромом дефицита Glut1 в большинстве случаев не выявляются патологические изменения. В ряде случаев описаны: гиперинтенсивный сигнал от U-образных волокон, расширение периваскулярных пространств Вирхова-Робина, задержка миелинизации [71, 74].
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 проведение ультразвукового исследования (УЗИ) органов брюшной полости (комплексного) и ультразвуковое исследование почек для оценки исходного состояния внутренних органов перед введением в кетогенную диету, а также их дальнейшего мониторинга [89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: УЗИ органов брюшной полости и почек проводится с целью исключения структурных аномалий, определения состояния внутренних органов перед началом кетогенной диеты, а также последующего мониторинга. Исследование проводится до введения в КД, через 3 месяца и далее 1 раз в 6 месяцев [69, 72].
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 регистрация электрокардиограммы для оценки частоты и регулярности сердечных сокращений, внутрисердечной проводимости перед введением в кетогенную диету, а также их дальнейшего мониторинга [89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: исследование проводится до введения в КД, через 3 месяца и далее 1 раз в 6 месяцев.
2.5 Иные диагностические исследования
Частота проведения обследований подробно описана в Приложении Г3.
Консультации специалистов могут оказываться пациентам на разных этапах оказания медицинской помощи, в том числе в период диагностики заболевания.
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 прием (осмотр, консультация) врача-генетика первичный и повторный для проведения дифференциальной диагностики, выбора тактики ДНК-диагностики и интерпретации полученных результатов молекулярно-генетических методов исследования [18].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 прием (осмотр, консультация) врача-педиатра/врача-терапевта/врача общей практики первичный и повторный для исследования общего состояния здоровья пациентов [18].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 прием (осмотр, консультация) врача-невролога первичный и повторный для исследования неврологического статуса, назначения лечения и дополнительного обследования при необходимости [18].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
- Рекомендуется всем пациентам с подозрением на синдром дефицита Glut1 и с установленным диагнозом синдром дефицита Glut1 участие в диагностике и лечении мультидисциплинарной команды специалистов ввиду того, что заболевание может поражать различные органы и системы [92].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Всем пациентам с синдромом дефицита Glut1 проводятся приемы (осмотры) врача-педиатра/врача-терапевта/врача общей практики (семейного врача) (первичные и повторные) при диагностике и в динамическом наблюдении с целью оценки общего состояния и эффективности проводимой терапии, участия в назначении и проведении лечебно-реабилитационных мероприятий.
Всем пациентам с синдромом дефицита Glut1 проводятся приемы (осмотры, консультации) врача-невролога с соответствующей подготовкой и врача-генетика (первичные и повторные) для установления диагноза, уточнения тяжести поражения центральной нервной системы (ЦНС) и определения тактики лечебно-реабилитационных мероприятий [18].
Всем пациентам с синдромом дефицита Glut1 проводятся приемы (осмотры, консультации) врача-диетолога (первичные и повторные) для назначения кетогенной диеты и далее при необходимости с целью коррекции диетотерапии, осуществления контроля за ее соблюдением [3, 22, 79].
Пациентам с подозрением на синдром дефицита Glut1 проводится прием (осмотр, консультация) врача-генетика для установления диагноза; пациентам с установленным диагнозом синдром дефицита Glut1, семьям, имеющим родственников с синдромом дефицита Glut1 - для планирования деторождения [18].
При подозрении/наличии у пациента патологии органа зрения проводятся приемы (осмотры, консультации первичные и повторные) врача-офтальмолога [3, 62, 63].
При подозрении/наличии у пациента патологии сердечно-сосудистой системы проводятся приемы (осмотры, консультации первичные и повторные) врача-кардиолога/врача-детского кардиолога [56].
Для планирования индивидуальной реабилитационной программы психолого-педагогической поддержки пациентам и их семьям проводятся прием (тестирование, консультация) медицинского психолога и логопеда-дефектолога (первичные и повторные) [3, 80].
Консультации врача-физиотерапевта, врача-гастроэнтеролога, врача-нефролога, врача-анестезиолога-реаниматолога, врача по паллиативной медицинской помощи, врача-стоматолога/врача-стоматолога детского и других специалистов - по показаниям.
3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
Лечение синдрома дефицита Glut1 включает как патогенетическое лечение - диетотерапию, так и проведение симптоматической терапии, направленной на уменьшение вторичных осложнений. Ведение пациентов с синдромом дефицита Glut1 предполагает мультидисциплинарный подход с обязательным участием врачей-неврологов, врачей-генетиков, врачей-гастроэнтерологов и врачей других специальностей, имеющих опыт в лечении этого редкого заболевания.
3.1 Патогенетическое лечение
В настоящее время единственным эффективным средством лечения пациентов с синдромом дефицита Glut1 является кетогенная диета (КД).
- Рекомендуется всем пациентам с синдромом дефицита Glut1 назначение КД с использованием специализированных продуктов детского диетического лечебного питания при лекарственно-резистентной эпилепсии и других состояниях, при которых показана КД, жировых эмульсий, содержащих среднецепочечные триглицериды, с целью восстановления метаболических процессов в ЦНС и уменьшения клинических проявлений заболевания [26]
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: При КД метаболизм сдвигается в сторону расщепления жиров, и головной мозг начинает использовать альтернативные источники энергии - кетоновые тела (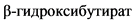 , ацетоацетат).
, ацетоацетат).
Кетоновые тела поступают в мозг, используя облегченную диффузию при помощи переносчика монокарбоксилата 1 (monocarboxylate transporter 1, MCT1) и обеспечивают альтернативный энергетический метаболизм в ЦНС.
КД в большинстве случаев хорошо переносится и позволяет уменьшить основные клинические проявления заболевания путем обеспечения контроля за эпилептическими приступами, редукции эпилептиформной активности на ЭЭГ, улучшения речевых, когнитивных и моторных функций. Однако приступы могут сохраняться даже на фоне соблюдения КД, а при купировании судорожного синдрома у пациентов иногда отмечается психоневрологический дефицит [54].
Неврологический исход во многом зависит от возраста, в котором начато лечение: больные, начавшие получать эффективное лечение в более раннем возрасте, имеют более благоприятный прогноз [39].
Специализированные продукты детского диетического лечебного питания при лекарственно-резистентной эпилепсии и других состояниях, при которых показана КД, могут использоваться для кормления тяжелобольных пациентов с года через рот и/или назогастральный зонд, через гастростому. Продукты и масла, используемые при КД, представлены в приложении Г16.
3.1.1 Рекомендации по проведению кетогенной диеты
КД представляет собой диету с высоким содержанием жиров и резко сниженным содержанием углеводов при контролируемом количестве белка, способствующую формированию и поддержанию в организме состояния кетоза.
Для достижения терапевтического эффекта требуется достижение уровня кетоновых тел в крови в интервале 2 - 6 ммоль/л. Пациенты с синдромом дефицита Glut1, особенно в первые годы жизни, для достижения клинического эффекта нуждаются в поддержании высокого уровня кетоновых тел на уровне верхней границы рекомендуемого терапевтического коридора (не менее 4 ммоль/л). В связи с этим исходным вариантом диетотерапии для них служит классический вариант КД.
В классической КД жировой компонент представлен натуральными животными жирами и растительными маслами. Традиционным кетогенным соотношением (отношение по массе жиров к белкам и углеводам) для классической КД служит 4:1 или 3:1. Однако точное значение определяется в каждом случае индивидуально с учетом уровня кетонемии. В приложении Г11 представлены подготовительный, начальный и основной этапы лечения на основе КД у детей раннего возраста.
Возможно включение в кетогенный рацион среднецепочечных триглицеридов (MCT - medium-chain triglycerides), которые способны обеспечить более высокий уровень кетонемии и одновременно снизить нагрузку на ферментные системы желудочно-кишечного тракта (ЖКТ). При этом диеты с MCT труднее переносятся детьми раннего возраста. Сравнительная характеристика КД, применяемых в лечении GLUT1 представлена в приложении Г6. В приложении Г5 представлено соотношение макронутриентов при различных вариантах КД, рассчитанных на рацион 1200 ккал.
- Рекомендуется пациентам с синдромом дефицита Glut1 при проблемах с переносимостью КД, а также подросткам и взрослым пациентам соблюдение модифицированной диеты Аткинса (МДА), допускающей более низкие значения кетогенного соотношения (1:1 - 2:1) с целью восстановления метаболических процессов в ЦНС и уменьшения клинических проявлений заболевания [81, 94, 95, 99].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: МДА не требует строгого контроля за количеством потребляемой пищи. Однако в качестве первоначального варианта при синдроме дефицита Glut1 следует использовать классический вариант КД.
- Рекомендуется детям первого года жизни, раннего, дошкольного и младшего школьного возраста с синдромом дефицита Glut1 соблюдение классической КД с целью обеспечения адекватного кетоза и восстановления метаболических процессов в ЦНС [26, 69].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Классической КД следует придерживаться как можно дольше, поскольку она способна обеспечить оптимальный уровень кетоновых тел в крови. МДА может быть предложена подросткам и взрослым как альтернатива классической КД, при появлении проблем с переносимостью в течение длительного времени. Диета с низким гликемическим индексом (ДсНГИ) не способна обеспечить стойкую кетонемию и в лечении синдрома дефицита Glut1 не используется.
Организация КД при синдроме дефицита Glut1 проводится в соответствии с общими требованиям к кетогенной диетотерапии [69]. Назначению КД должно предшествовать проведение комплексного обследования, включающего оценку неврологического, соматического и нутритивного статуса пациента, с использованием антропометрических, лабораторных и инструментальных исследований, а также анализ фармакотерапии. По результатам обследования принимается решение об инициации диетотерапии, разрабатывается индивидуальный план лечения и кетогенный рацион [72].
Показания и противопоказания к назначению КД представлены в приложении Г4.
Согласно результатам последних исследований диапазон применяемых соотношений КД составляет 2 - 4,5:1. Кетогенное соотношение определяется индивидуально для каждого пациента с учетом особенностей его кетогенеза и переносимости [2, 14, 24, 26, 32, 50].
- Рекомендуется всем пациентам с синдромом дефицита Glut1, соблюдающим КД, индивидуальный расчет калорийности диеты с целью обеспечения достаточных энергетических ресурсов для обеспечения адекватного физического развития и поддержания необходимого уровня кетоза [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Энергетическая ценность кетогенной диеты рассчитывается индивидуально для каждого пациента с учетом возраста, уровня двигательной активности и особенностей метаболизма.
При использовании КД калорийность часто ограничивается до 80 - 90% от суточной нормы здорового населения; однако доказанной эффективности такого расчета нет и большинство экспертов предпочитают не ограничивать поступление калорий [69].
Для определения потребности в энергии можно ориентироваться на среднесуточные нормы физиологических потребностей в энергии для здоровых детей согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17 - 24 okt 2001), представленные в приложении Г7 и Г10.
Детям со значительным ограничением двигательной активности для расчета энергетической потребности используется индивидуальный расчет потребности в энергии с использованием уравнения Шофилда [82] (приложение Г15).
Может быть предложено равномерное деление рассчитанного количества калорий в течение дня, можно организовать основные приемы пищи и дополнительные, меньшей энергетической ценности и, соответственно, с меньшим количеством углеводов.
Об адекватном поступлении калорий свидетельствуют темпы прибавки массы тела и уровень кетоновых тел.
Поступление избыточного количества энергии сопряжено с высокими прибавкам массы тела и снижением кетоза, в то время как недостаток поступления калорий может стать причиной задержки физического развития и избыточной кетонемии.
Коррекция энергетической ценности рациона осуществляется на протяжении всей диетотерапии с учетом динамики массо-ростовых показателей [72].
Оценка физического развития производится с учетом идеального соотношения вес/возраст и вес/рост.
Антропометрические исследования проводится детям первого года жизни - еженедельно, детям 1 - 2-х лет жизни - 1 раз в две недели, детям с 3-х лет - ежемесячно.
- Рекомендуется всем пациентам с синдромом дефицита Glut1, соблюдающим КД, индивидуальный расчет потребности в белках, основанный на возрастных рекомендациях с целью обеспечения адекватных параметров физического развития [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Количество белка в рационе не должно быть ниже безопасного уровня потребления. Для расчета потребности в белке больным с GLUT 1 следует использовать значения RDA (recommended dietary allowances - рекомендуемые нормы питания), представленные в приложении Г8 и Г10 [18, 58].
При классической КД на долю белка в среднем приходится 5 - 15% от общей калорийности. При МДА количество белка достаточно высоко и составляет до 30% от энергоценности.
При задержке роста для достижения его адекватных темпов уровень потребления белка корректируется в сторону увеличения.
- Рекомендуется всем пациентам с синдромом дефицита Glut1, соблюдающим КД, индивидуальный расчет потребности в жирах с учетом энергетической ценности рациона с целью обеспечения заданного кетогенного соотношения [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Классическая КД основана преимущественно на длинноцепочечных триглицеридах (LCT - long chain triglyceride) и в качестве источника жиров предполагает использование натуральных животных жиров и растительных масел. С растительными маслами должно поступать не менее 60% жиров. В зависимости от индивидуальной переносимости и особенностей кетогенеза кетогенное соотношение может значительно варьировать от уровня 2:1 до 4,5:1, соответственно доля энергии от потребления жиров составляет 70 - 90%.
При MCT-диете 30 - 60% жиров представлено среднецепочечными триглицеридами. MCT обладают способностью стимулировать кетогенез, поэтому допускаются более низкие значения кетогенного соотношения (до 1:1) и, соответственно, большее количество углеводов. При этом у детей младших возрастных групп MCT-диета часто сопровождается явлениями непереносимости со стороны ЖКТ, в связи с чем она не назначается детям первого года жизни и раннего возраста. В качестве источника MCT используются специальные жировые модули, а также кокосовое масло (45% жиров которого представлено MCT). Включение специальных модулей более оправдано, учитывая высокое содержание в кокосовом масле насыщенных жиров. Различия в клинической эффективности классической КД и MCT-диеты не установлены [5]. При необходимости включения в рацион питания MCT используются во все приемы пищи для снижения возможных побочных эффектов [72].
Масло с MCT также используется в качестве компонента классической КД с целью усиления кетогенеза, снижения нагрузки на ЖКТ, а также стимуляции моторики кишечника. Общее количество MCT, не вызывающее проблем с переносимостью в раннем возрасте, не превышает 10 - 25% от суточной калорийности [39].
Для МАД количество жиров составляет 60 - 65% энергоценности.
- Рекомендуется всем пациентам с синдромом дефицита Glut1, соблюдающим КД, расчет потребности в углеводах в зависимости от варианта КД и энергетической ценности рациона питания с целью обеспечения адекватного кетогенного соотношения [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: В классической КД количество углеводов рассчитывается после определения суммарного количества белков и углеводов и вычетом количества необходимого белка. При расчете количества углеводов, учитывают все возможные источники, в том числе присутствующие в фармпрепаратах. Количество углеводов рассчитывается на каждый прием пищи, исходя из его вклада в общую энергоценность.
Для МДА допустимое количество углеводов в сутки составляет 10 - 15% от энергоценности. Рекомендуется постепенное достижение необходимого количества углеводов в рационе, начиная с 10 грамм в сутки.
Допускается применение подсластителей в регламентированных количествах [39].
Количество углеводов рассчитывается на каждый прием пищи, исходя из его вклада в общую энергоценность рациона. Может быть предложено равномерное деление в течение дня, можно организовать основные приемы пищи и ввести дополнительный с меньшей энергетической ценностью и, соответственно, с меньшим количеством углеводов.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 избегать приема углевод-содержащих лекарственных препаратов с целью поддержания адекватного кетогенного соотношения [92].
Уровень убедительности рекомендации C (уровень достоверности доказательств - 5)
Комментарии: по возможности, такие препараты необходимо заменять альтернативными вариантами без включения углеводов.
- Рекомендуется всем пациентам с синдромом дефицита Glut1, соблюдающим КД, поддерживать достаточный питьевой режим с целью обеспечения адекватного водно-электролитного баланса [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Учитывая, что КД обладает диуретическим эффектом, необходимо обеспечить достаточное поступление жидкости с целью предотвращения гипогидратации и связанных с ней метаболических осложнений, в первую очередь со стороны почек (нефролитиаза), а также нарушений моторики ЖКТ (запоров).
При хорошей переносимости кетогенной диеты обычная потребность в жидкости рассчитывается по суточной калорийности (объем жидкости/сут = не менее ккал/сут, т.е. 1000 ккал/сут - не менее 1 л жидкости в сутки).
При повышенных потерях жидкости ее количество необходимо корректировать (гипертермия, рвота, диарея и др.).
Необходимое количество потребляемой жидкости в зависимости от возраста и веса представлено в приложении Г9.
- Рекомендуется пациентам до 3-х лет с синдромом дефицита Glut1 осуществлять введение в классическую КД в условиях стационара в связи с необходимостью квалифицированного медицинского наблюдения за пациентами [93, 96].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: Введение КД детям первых 3-х лет жизни осуществляется исключительно в стационаре. МДА может вводиться в амбулаторных условиях.
Введение кетогенного рациона осуществляется различными способами. Для быстрого достижения терапевтического уровня кетоновых тел может быть использована короткая голодная пауза (не превышающая 6 часов), за которой следует дробное (в течение 2 - 3 дней) введение кетогенного рациона, начиная с 1/2 необходимой энергетической ценности. Также возможно введение КД заменяя по 1 приему пищи в день на кетогенный каждые 1 - 2 дня. Реже используется прогредиентное повышение кетогенного соотношения рациона от 1:1 (или 2:1) до достижения необходимого уровня кетонемии. Соотношение изменяется в зависимости от состояния и самочувствия ребенка каждые 3 - 4 дня или еженедельно. К недостаткам последнего варианта относится достижение терапевтического коридора кетоновых тел вне стационара, что нежелательно, учитывая возможность индивидуальных негативных реакций на кетоз.
3.2 Симптоматическая терапия - противопилептические препараты
Противоэпилептические препараты (ПЭП), как правило, неэффективны или дают незначительное улучшение при отсутствии лечения с помощью кетогенной диеты. Некоторые ПЭП могут иметь относительные противопоказания при назначении в качестве сопутствующей терапии у детей, находящихся на кетогенной диете.
- Рекомендуется пациентам с синдромом дефицита Glut1 при неполной эффективности КД назначение ПЭП с целью снижения частоты эпилептических приступов (в соответствии с клиническими рекомендациями по эпилепсии) [83].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Назначение ПЭП целесообразно при недостаточной эффективности патогенетической терапии - кетогенной диеты.
Применение ряда противоэпилептических препаратов ассоциируется с повышением частоты возникновения побочных эффектов КД и/или снижением ее эффективности.
Прием ингибиторов карбоангидразы - ацетазоламида**, топирамата**, сультиама может значительно усиливать ацидоз. Топирамат**, ацетазоламид** и зонисамид повышают риск уролитиаза. Снижение эффективности диетотерапии отмечено на фоне приема ламотриджина. Прием вальпроевой кислоты** повышает риск гепатотоксичности [26].
3.3 Сопутствующая терапия на фоне применения кетогенной диеты
Витамины и минеральные добавки
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на КД, назначение поливитаминов в комбинации с минеральными веществами с обязательным присутствием комплекса витаминов группы B, препаратов селена, препаратов кальция, препаратов калия, магния и дополнительная дотация витамина D и его аналогов в возрастной дозировке с целью предотвращения дефицита нутриентов [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Рацион КД не является физиологическим и полноценным, в связи с этим нуждается в дополнении поливитаминами и минеральными добавками в форме, не содержащей углеводов.
Из-за ограничения в рационе фруктов, овощей, зерновых и продуктов, богатых кальцием, пациентам, находящимся на КД, назначают поливитамины с минеральными веществами, не содержащие углеводы. Учитывая, что продукты, используемые при КД, как правило, содержат недостаточное количество витамина D и кальция, а у детей с фармакорезистентной эпилепсией существует риск наличия дефицита витамина D еще до начала терапии КД показано добавление витамина D и его аналогов в соответствии с рекомендуемой возрастной нормой. Часть экспертов сошлись во мнении в назначении повышенных доз витамина D и его аналогов сверх рекомендаций RDA [69].
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на КД, прием левокарнитина с целью восполнения его уровня [91].
Уровень убедительности рекомендации B (уровень достоверности доказательств - 3)
Комментарии: Дети раннего возраста, пациенты, получающие фармакотерапию вальпроевой кислотой**, имеющие дефицит массы тела и длительно соблюдающие КД имеют повышенный риск формирования вторичного дефицита карнитина. Дефицит карнитина может проявляться утомляемостью и мышечной слабостью, но при этом редко приводит к нарушению работы печени и сердечно-сосудистой системы. Учитывая необходимость длительного соблюдения КД при синдроме дефицита Glut1, пациенты относятся к группе риска по вторичному дефициту карнитина и нуждаются в его дотации в дозировке 50 - 100 мг/кг/день [107].
- Рекомендуется пациентам старше 12 лет с синдромом дефицита Glut1, находящимся на КД, назначение G04B Препаратов для лечения урологических заболеваний под контролем pH мочи с целью предотвращения риска развития нефролитиаза [69, 72].
Уровень убедительности рекомендации B (уровень достоверности доказательств - 3)
Комментарии: стоит учитывать повышение риска возникновения мегалобластной анемии на фоне применения данной группы препаратов за счет снижения абсорбции фолиевой кислоты [69].
3.4 Обследования перед введением в кетогенную диету
Необходимые обследования перед введением в КД описаны в приложении Г3. Рекомендуемый контрольный клинический мониторинг при соблюдении КД представлен в приложениях Г12 и Г13.
3.4.1 Лабораторные исследования на начальном этапе кетогенной диеты
- Рекомендуется пациентам с синдромом дефицита Glut1 проведение общего (клинического) анализа крови развернутого перед началом КД и, далее через 3 и 6 месяцев после начала КД, далее - 1 раз в 6 месяцев с целью мониторинга состояния и своевременного выявления возможных противопоказаний [69, 70, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: проводят исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула) и исследование скорости оседания эритроцитов. Атипичные варианты течения в редких случаях могут сопровождаться гемолитической анемией. Исследование проводится в процессе динамического наблюдения, частота определяется индивидуально.
- Рекомендуется пациентам с синдромом дефицита Glut1 проведение общего (клинического) анализа мочи перед началом КД и далее через 3 и 6 месяцев после начала КД, далее - 1 раз в 6 месяцев с целью мониторинга состояния и своевременного выявления возможных противопоказаний [69, 70, 106].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: необходимы визуальное исследование мочи - цвет, прозрачность, определение концентрации водородных ионов (pH) мочи, определение удельного веса (относительной плотности) мочи, определение белка в моче, исследование уровня глюкозы в моче, обнаружение кетоновых тел в моче, желчных пигментов в моче, исследование уровня билирубина в моче, обнаружение гемоглобина в моче, исследование уровня нитритов в моче, микроскопическое исследование осадка мочи - эпителия, лейкоцитов, эритроцитов, кристаллов солей, бактерий, слизи. Исследование проводится при постановке диагноза и далее в процессе динамического наблюдения, частота определяется индивидуально.
- Рекомендуется проведение анализа крови биохимического общетерапевтического пациентам с синдромом дефицита Glut1 перед началом КД и, далее, через 6 недель после начала КД, через 3 и 6 месяцев, далее - 1 раз в 6 месяцев с целью мониторинга состояния и своевременного выявления возможных противопоказаний [3, 69, 70, 72, 77].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: при синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать состояние внутренних органов. Необходимо провести исследование уровня общего белка в крови, исследование уровня альбумина в крови, исследование уровня мочевины в крови, исследование уровня креатинина в крови, исследование уровня триглицеридов в крови, исследование уровня холестерина в крови, исследование уровня холестерина липопротеинов низкой плотности, исследование уровня липопротеинов в крови, определение активности щелочной фосфатазы в крови, исследование уровня общего билирубина в крови, исследование уровня свободного и связанного билирубина в крови, исследование уровня 25-OH витамина Д в крови, исследование уровня глюкозы в крови, определение активности аспартатаминотрансферазы в крови, определение активности аланинаминотрансферазы в крови, определение активности амилазы в крови, исследование уровня натрия в крови, исследование уровня калия в крови, исследование уровня общего кальция в крови, исследование уровня общего магния в сыворотке крови.
- Рекомендуется пациентам с синдромом дефицита Glut1 проведение Исследование уровня кальция, уровня мочевой кислоты, уровня креатинина в моче перед началом КД, потом через 3 и 6 месяцев, далее - 1 раз в 6 месяцев с целью мониторинга состояния и своевременного выявления возможных противопоказаний [3, 69, 70, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: при синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать состояние внутренних органов.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 обнаружение кетоновых тел в моче или обнаружение кетоновых тел в моче экспресс-методом для оценки их исходного уровня перед введением в кетогенную диету [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: При синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать состояние внутренних органов.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 и подтвержденным синдромом дефицита Glut1 обнаружение кетоновых тел в крови с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования для оценки эффективности кетогенной диеты - определение исходного уровня перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Кетоны можно измерять в крови (наиболее информативно) или в моче. На начальном этапе диеты рекомендуется измерение кетонов в крови с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования, поскольку этот метод более точен и не подвержен эффекту разведения мочи или влиянию любых возможных изменений водного гомеостаза, которые могут отмечаться у младенцев первых месяцев жизни. Кетоны крови следует измерять дважды в день, собирая образец из пальца или пятки. Целевое значение кетонов в крови на КД 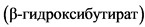 - 2 - 6 ммоль /л, в моче (ацетоацетат) - 8 - 16 ммоль/л [70].
- 2 - 6 ммоль /л, в моче (ацетоацетат) - 8 - 16 ммоль/л [70].
Для оценки эффективности КД необходим регулярный самоконтроль уровня кетоновых тел в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня кетонов.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 и подтвержденным синдромом дефицита Glut1 исследование уровня глюкозы в крови с помощью системы мониторинга глюкозы ИВД для домашнего использования или системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования для оценки эффективности кетогенной диеты - перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Для оценки эффективности КД необходим регулярный самоконтроль уровня глюкозы в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня глюкозы.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 на начальном этапе КД определение кислотно-щелочного состояния и газов крови (исследование уровня водородных ионов (pH) крови, исследование уровня буферных веществ в крови), уровня глюкозы в крови, исследование уровня натрия в крови, уровня общего кальция в крови, уровня калия в крови, уровня хлоридов в крови, исследование уровня молочной кислоты в крови с целью диагностики метаболических нарушений [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: на этапе введения кетогенной диеты данные параметры оцениваются 1 раз в 1 - 3 дня, далее при стабильном кетозе по показаниям.
3.4.2 Инструментальные исследования перед началом терапии кетогенной диетой
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение электроэнцефалографии с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: ЭЭГ должна быть проведена как минимум за 3 месяца до назначения КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение электроэнцефалографии с видеомониторингом до еды, во время и после приема пищи с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности при невозможности проведения стандартной (рутинной) электроэнцефалографии [73].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3)
Комментарии: ЭЭГ с видеомониторингом должна быть проведена при невозможности проведения стандартной (рутинной) ЭЭГ как минимум за 3 месяца до начала КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
У пациентов с Glut1 интериктальная ЭЭГ обычно в норме. По результатам ЭЭГ чаще всего определяются мультифокальные спайк-волновые разряды, полиспайк-волновые разряды и билатерально синхронные.
У младенцев чаще регистрируется региональное замедление и эпилептиформные разряды. У детей в возрасте двух лет и старше наблюдается генерализованная спайк-волна с частотой 2,5 - 4 Гц [27, 31, 74].
Может наблюдаться любой тип приступов, чаще встречаются генерализованные тонические или клонические, абсансы, фокальные, миоклонические, атонические.
Важным патогномоничным признаком заболевания является регресс эпилептических приступов и патологических изменений по ЭЭГ после приема пищи.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение магнитно-резонансной томографии головного мозга (МРТ) головного мозга для определения степени тяжести заболевания и дифференциальной диагностики с нейродегенеративными и другими заболеваниями [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Комбинированный эндотрахеальный наркоз или комбинированный ингаляционный наркоз по показаниям - применение пропофола** детям старше 1 месяца жизни - с осторожностью (см. подраздел 3.6) [75]. Магнитно-резонансная томография проводится с целью исключения структурных аномалий головного мозга и нейрометаболических заболеваний. При нейровизуализации у пациентов с синдромом дефицита Glut1 в большинстве случаев не выявляются патологические изменения. В ряде случаев описаны: гиперинтенсивный сигнал от U-образных волокон, расширение периваскулярных пространств Вирхова-Робина, задержка миелинизации [71, 74].
- Рекомендуется проведение ультразвукового исследования почек пациентам с синдромом дефицита Glut1 на КД с целью мониторинга состояния и своевременного выявления возможных противопоказаний [69, 70, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: также при необходимости проводят ультразвуковое исследование органов брюшной полости (комплексное) и почек проводится с целью исключения структурных аномалий, определения состояния внутренних органов перед началом кетогенной диеты, а также последующего мониторинга. Исследование проводится до введения в КД, через 3 месяца и далее 1 раз в 6 месяцев (чаще - по показаниям) [69, 72].
- Рекомендуется всем пациентам с синдромом дефицита Glut1 регистрация электрокардиограммы для оценки частоты и регулярности сердечных сокращений, внутрисердечной проводимости перед введением в кетогенную диету, а также их дальнейшего мониторинга [69, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: исследование проводится до введения в КД, через 3, 6 месяцев и далее 1 раз в 6 месяцев.
3.5 Терапия побочных эффектов и осложнений кетогенной диеты
Таблица возможных побочных эффектов при соблюдении КД, а также их профилактика и коррекция представлена в приложении Г14.
- Рекомендуется пациентам с синдромом дефицита Glut1 в случае гиперкетонемии прием углеводов с целью снижения уровня кетонов в крови [71, 77].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 5)
Комментарии: Появление одышки, тахикардии, нарастание сонливости или спутанность сознания, повторная рвота может служить проявлением гиперкетонемии (диагностируется при повышении уровня кетонов в крови свыше 6 ммоль/л). Тактика заключается в приеме 5 г углеводов и повторном мониторинге кетоновых тел в крови через 15 - 20 минут. При необходимости прием углеводов следует повторить.
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на КД, в случае повышения кетонов в крови (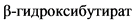 выше 6 ммоль/л), в моче (ацетоацетат выше 16 ммоль/л) и появлении признаков ацидоза (слабость, тошнота, фруктовый (кетоновый) запах в выдыхаемом воздухе, тахикардия, тахипноэ) пероральный прием углеводов в дозе 2 - 5 г с целью снижения уровня кетонов до целевых значений (2 - 6 ммоль/л в крови, 8 - 16 ммоль/л в моче) [71, 77].
выше 6 ммоль/л), в моче (ацетоацетат выше 16 ммоль/л) и появлении признаков ацидоза (слабость, тошнота, фруктовый (кетоновый) запах в выдыхаемом воздухе, тахикардия, тахипноэ) пероральный прием углеводов в дозе 2 - 5 г с целью снижения уровня кетонов до целевых значений (2 - 6 ммоль/л в крови, 8 - 16 ммоль/л в моче) [71, 77].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Необходимо измерить уровень кетонов через 15 - 20 минут после лечения, если результатов нет - повторить лечение.
При снижении уровня глюкозы ниже 3 ммоль/л необходимо связаться с врачом. Следует дать ребенку 2 - 5 г углеводов (50 мл фруктового сока или 100 мл молока).
Если эпизоды избыточного кетоза случается часто, следует рассмотреть вопрос о перерасчете кето-соотношения.
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на кетогенной диете, в случае возникновения гипогликемии (уровень глюкозы ниже 2,6 ммоль/л) пероральный прием углеводов в дозе 5 г с целью коррекции гипогликемии [77, 100].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Провести исследование уровня глюкозы в крови через 15 - 20 минут после лечения, если результатов нет - повторить лечение.
Если пероральный прием углеводов невозможен, у пациента судороги или он находится без сознания, рекомендовано внутривенное введение 10% р-ра декстрозы** в дозе 5 мл/кг [77].
Если гипогликемия случается часто - рассмотреть вопрос о перерасчете кето-соотношения.
- Рекомендуется исследование уровня глюкозы в крови пациентам до 18 лет с синдромом дефицита Glut1, находящимся на КД, в случае развития метаболического ацидоза с целью определения причины сдвига кислотно-основного состояния [76]
Уровень убедительности рекомендации C (уровень достоверности доказательств - 4)
Комментарии: также исследуют уровень кетонов в крови. Если метаболический ацидоз связан с гипогликемией или избыточным кетозом, лечение натрия гидрокарбонатом**, как правило, не требуется. Достаточно скорректировать уровень глюкозы и кетонов в крови.
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на КД, в случае развития метаболического ацидоза, не связанного с гипогликемией или избыточным кетозом, введение растворов, влияющих на водно-электролитный баланс, с целью коррекции метаболических нарушений [84].
Уровень убедительности рекомендации C (уровень достоверности доказательств - 5)
Комментарии: для коррекции дегидратации, интоксикации используются растворы натрия гидрокарбоната** (код АТХ - B05XA), Калия хлорид + Натрия гидрокарбонат + Натрия хлорид. Натрия гидрокарбонат** применяется в виде 8,4% и 4,2% раствора для удобства перерасчета на ммоль NaHCO. Его дозировка (ммоль) определяется по формуле: (-BE) x масса тела (кг) X 0,3. Кроме того, пациентам рекомендуется щелочное питье - раствор соды из расчета 1/2 - 1 чайная ложка на 200 мл воды, щелочные минеральные воды.
В зависимости от тяжести состояния каждые 6 - 12 часов контролировать показатели кислотно-основного состояния крови, уровня натрия и калия в крови.
Рассмотреть вопрос о снижении дозы или отмене ингибиторов карбоангидразы (топирамат**, зонисамид и ацетазоламид**), если они были назначены ранее.
3.6 Взаимодействие кетогенной диеты с лекарственными препаратами.
- Рекомендуется пациентам с синдромом дефицита Glut1, находящимся на КД, ограничить назначение лекарственных препаратов, содержащих углеводы, и пищевых добавок в связи с повышением риска снижения кетонемии [69, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: При необходимости назначения данных препаратов, рекомендовано учитывать количество углеводов, содержащихся в препаратах, при расчете допустимого количества углеводов с целью обеспечения адекватной кетонемии.
4. Медицинская реабилитация и санаторно-курортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
Специфической реабилитации пациентам с дефицитом Glut1 не требуется. В круг реабилитационных мероприятий могут быть включены занятия с медицинским психологом, отдых в специализированных санаториях, а также клинико-психологическая адаптация, социально-реабилитационная работа с участием специалистов и социальных работников, курсы общего массажа медицинского. Важно предотвратить осложнения и способствовать школьной и профессиональной интеграции, общению со сверстниками.
- Рекомендованы пациентам с синдромом дефицита Glut1 приемы (консультации, тестирование) (первичные и повторные) медицинского психолога, разработка индивидуальной программы дефектологической реабилитации, нейропсихологические коррекционно-восстановительные процедуры, клинико-психологическое консультирование при нарушениях психических функций, нейропсихологическая реабилитация, школа психологической профилактики/реабилитации для пациентов и родственников для коррекции психологического состояния при наличии показаний [39, 97, 98].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: У пациентов с синдромом дефицита Glut1 наблюдается задержка психоречевого развития, поведенческие нарушения, требующие комплексного наблюдения и терапии медицинских психологов, дефектологов, врачей физической и реабилитационной медицины/врачей по медицинской реабилитации или исполняющих их функции.
Психолого-педагогическая реабилитация детей подросткового возраста. Они сталкиваются с проблемами периода полового созревания, которые сопровождается стремлением к независимости, протестным поведением, а также подверженностью влияния сверстников. Это период частых отказов пациентов подростков от диетотерапии, употребления продуктов специализированного питания, а также высокого риска потери метаболического контроля и полной утраты медицинского наблюдения [97].
5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
5.1 Профилактика
- Рекомендуется пациентам с синдромом дефицита Glut1 или его официальным представителям прием (осмотр, консультация) врача-генетика после установления диагноза для получения информации о генетическом риске, обсуждения возможностей пренатальной и преимплантационной диагностики [5, 14].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Пациентам с синдромом дефицита Glut1 показано медико-генетическое консультирование с целью определения генетического прогноза для семьи.
В большинстве случаев заболевание возникает вследствие гетерозиготной мутации в гене SLC2A1, возникающей преимущественно de novo. Реже (примерно в 10% описанных случаев) происходит наследование по аутосомно-доминантному типу, при этом у родителя с выявленной патогенной мутацией SLC2A1, возможна легкая форма заболевания. Возможным объяснением более мягкого фенотипа может быть сниженная активность белкового продукта вследствие тканевого мозаицизма [2]. Имеются также сообщения о редком аутосомно-рецессивном наследовании заболевания.
- Рекомендуется детям с синдромом дефицита Glut1 проведение вакцинации в соответствии с национальным календарем с целью предотвращения вакциноуправляемых инфекций [105].
Уровень убедительности доказательств C (уровень достоверности рекомендации - 4)
Комментарии: противопоказания определяются индивидуально.
Синдром дефицита Glut1 не является противопоказанием к проведению вакцинации другими вакцинами.
5.2 Диспансерное наблюдение
Контроль за динамикой показателей исходного обследования:
- для детей первого года жизни: через 1 - 1,5 месяца после начала терапии КД, далее через 3 мес., 6 мес, 12 мес, далее 1 раз в 6 - 12 мес.
- для детей старше 2-х лет: через 3 мес, 6 мес, 12 мес, далее 1 раз в 6 - 12 мес. [69, 72].
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение общего (клинического) анализа крови развернутого (исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула) и исследование скорости оседания эритроцитов) для оценки основных параметров кроветворения и наличия воспалительных процессов перед началом КД через 3 - 6 - 12 месяцев после начала КД, далее - 1 раз в 6 месяцев [69, 70, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Атипичные варианты течения в редких случаях могут сопровождаться гемолитической анемией. Исследование проводится в процессе динамического наблюдения, частота определяется индивидуально.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 проведение общего (клинического) анализа мочи для оценки общих свойств мочи и наличия воспалительных процессов перед началом КД, через 3 - 6 - 12 месяцев, далее - 1 раз в 6 месяцев [69, 70, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: необходимы визуальное исследование мочи - цвет, прозрачность, определение концентрации водородных ионов (pH) мочи, определение удельного веса (относительной плотности) мочи, определение белка в моче, исследование уровня глюкозы в моче, обнаружение кетоновых тел в моче, желчных пигментов в моче, исследование уровня билирубина в моче, обнаружение гемоглобина в моче, исследование уровня нитритов в моче, микроскопическое исследование осадка мочи - эпителия, лейкоцитов, эритроцитов, кристаллов солей, бактерий, слизи. Исследование проводится при постановке диагноза и далее в процессе динамического наблюдения, частота определяется индивидуально.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 проведение анализа крови биохимического общетерапевтического для оценки функций внутренних органов перед началом КД, через 3 - 6 - 12 месяцев, далее - 1 раз в 6 месяцев [3, 69, 72, 77].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: При синдроме дефицита Glut1 назначается кетогенная диета, при введении в которую, а также в процессе соблюдения необходимо оценивать состояние внутренних органов. Необходимо провести исследование уровня общего белка в крови, исследование уровня альбумина в крови, исследование уровня мочевины в крови, исследование уровня креатинина в крови, исследование уровня триглицеридов в крови, исследование уровня холестерина в крови, исследование уровня холестерина липопротеинов низкой плотности, исследование уровня липопротеинов в крови, определение активности щелочной фосфатазы в крови, исследование уровня общего билирубина в крови, исследование уровня свободного и связанного билирубина в крови, исследование уровня 25-OH витамина Д в крови, исследование уровня глюкозы в крови, определение активности аспартатаминотрансферазы в крови, определение активности аланинаминотрансферазы в крови, определение активности амилазы в крови, исследование уровня натрия в крови, исследование уровня калия в крови, исследование уровня общего кальция в крови, исследование уровня общего магния в сыворотке крови
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 проведение исследование уровня фосфора в моче, уровня кальция в моче, уровня мочевой кислоты в моче, уровня креатинина в моче, уровня калия в моче, уровня натрия в моче, кетоновых тел в моче, уровня хлоридов в моче для оценки функций внутренних органов перед началом КД через 3 - 6 - 12 месяцев после начала КД, далее - 1 раз в 6 месяцев [69, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
- Рекомендуется: пациентам с синдромом дефицита Glut1, находящимся на КД, исследование уровня 25-OH витамина Д в крови в крови 1 раз в 6 месяцев с целью своевременного выявления его дефицита и адекватной коррекции [69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 и подтвержденным синдромом дефицита Glut1 обнаружение кетоновых тел в крови с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования для оценки эффективности кетогенной диеты - определение исходного уровня перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Кетоны можно измерять в крови (наиболее информативно) или в моче. На начальном этапе диеты рекомендуется измерение кетонов в крови с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования, поскольку этот метод более точен и не подвержен эффекту разведения мочи или влиянию любых возможных изменений водного гомеостаза, которые могут отмечаться у младенцев первых месяцев жизни. Кетоны крови следует измерять дважды в день, собирая образец из пальца или пятки. Целевое значение кетонов в крови на КД 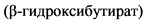 - 2 - 6 ммоль/л, в моче (ацетоацетат) - 8 - 16 ммоль/л [70].
- 2 - 6 ммоль/л, в моче (ацетоацетат) - 8 - 16 ммоль/л [70].
Для оценки эффективности КД необходим регулярный самоконтроль уровня кетоновых тел в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня кетонов.
- Рекомендуется всем пациентам с клиническими проявлениями синдрома дефицита Glut1 и подтвержденным синдромом дефицита Glut1 исследование уровня глюкозы в крови с помощью системы мониторинга глюкозы ИВД для домашнего использования или системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования для оценки эффективности кетогенной диеты - перед введением в кетогенную диету и далее 2 раза в сутки [39, 69, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Для оценки эффективности КД необходим регулярный самоконтроль уровня глюкозы в крови - 2 раза в сутки с помощью системы мониторинга глюкозы/кетонов в крови ИВД для домашнего использования (аппарат, ланцеты, тест-полоски). Тест-полоски должны быть совместимы с аппаратом для измерения уровня глюкозы.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение электроэнцефалографии с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: ЭЭГ должна быть проведена как минимум за 3 месяца до назначения КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение электроэнцефалографии с видеомониторингом до еды, во время и после приема пищи с целью выявления эпилептиформной активности, оценки степени тяжести заболевания перед назначением и в процессе КД для контроля ее эффективности при невозможности проведения стандартной (рутинной) электроэнцефалографии [73].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3)
Комментарии: ЭЭГ с видеомониторингом должна быть проведена при невозможности проведения стандартной (рутинной) ЭЭГ как минимум за 3 месяца до начала КД, далее - после достижения устойчивого уровня кетоза, далее - 1 раз в 6 месяцев.
У пациентов с Glut1 интериктальная ЭЭГ обычно в норме. По результатам ЭЭГ чаще всего определяются мультифокальные спайк-волновые разряды, полиспайк-волновые разряды и билатерально синхронные.
У младенцев чаще регистрируется региональное замедление и эпилептиформные разряды. У детей в возрасте двух лет и старше наблюдается генерализованная спайк-волна с частотой 2,5 - 4 Гц [27, 31, 74].
Может наблюдаться любой тип приступов, чаще встречаются генерализованные тонические или клонические, абсансы, фокальные, миоклонические, атонические.
Важным патогномоничным признаком заболевания является регресс эпилептических приступов и патологических изменений по ЭЭГ после приема пищи.
- Рекомендуется всем пациентам с синдромом дефицита Glut1 проведение магнитно-резонансной томографии головного мозга (МРТ) головного мозга для определения степени тяжести заболевания и дифференциальной диагностики с нейродегенеративными и другими заболеваниями [13].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: магнитно-резонансная томография проводится с целью исключения структурных аномалий головного мозга и нейрометаболических заболеваний. При нейровизуализации у пациентов с синдромом дефицита Glut1 в большинстве случаев не выявляются патологические изменения. В ряде случаев описаны: гиперинтенсивный сигнал от U-образных волокон, расширение периваскулярных пространств Вирхова-Робина, задержка миелинизации [71, 74].
- Рекомендуется проведение ультразвукового исследования почек пациентам с синдромом дефицита Glut1 для оценки исходного состояния внутренних органов перед введением в кетогенную диету, а также их дальнейшего мониторинга [69, 70, 72].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: также при необходимости проводят ультразвуковое исследование органов брюшной полости (комплексное), УЗИ органов брюшной полости и почек проводится с целью исключения структурных аномалий, определения состояния внутренних органов перед началом кетогенной диеты, а также последующего мониторинга. Исследование проводится до введения в КД, через 3 месяца и далее 1 раз в 6 месяцев (чаще - по показаниям) [69, 72].
- Рекомендуется всем пациентам с синдромом дефицита Glut1 регистрация электрокардиограммы для оценки частоты и регулярности сердечных сокращений, внутрисердечной проводимости перед введением в кетогенную диету, а также их дальнейшего мониторинга [69, 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: Исследование проводится до введения в КД, через 3 месяца и далее 1 раз в 6 месяцев.
- Рекомендуются пациентам с синдромом дефицита Glut1, находящимся на КД, для наблюдения применять мультидисциплинарный подход ввиду того, что заболевание характеризуется поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [3, 69, 72, 85].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5)
Комментарии: показаны первичные и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, врача-диетолога, врача-гастроэнтеролога, врача-эндокринолога, врача-кардиолога, врача-детского кардиолога, врача-нефролога, врача-педиатра/врача-терапевта/врача общей практики (семейного врача), а также врачей других специальностей пациентам с синдромом дефицита Glut1, находящимся на КД, имеющим нарушения функций соответствующих органов и систем
6. Организация оказания медицинской помощи
Пациентам с Glut1, в зависимости от необходимости, может быть оказана медицинская помощь любого вида, условий, форм, предусмотренных законодательством Российской Федерации.
Ведением пациентов с дефицитом Glut1 обычно занимается врач-невролог, врач-диетолог, врач-гастроэнтеролог, врач-педиатр/врач-терапевт, а также другие врачи-специалисты, имеющие опыт ведения пациентов с дефицитом Glut.
Чаще госпитализация осуществляется в неврологическое отделение.
Необходимые осмотры врачей-специалистов, лабораторные и инструментальные обследования и рекомендуемая частота их проведения представлены в разделе 5.2
Показания для плановой госпитализации:
- Проведение диагностики и лечения, требующие круглосуточного медицинского наблюдения;
- Состояние, требующее активного лечения и круглосуточного медицинского наблюдения;
- Состояние, требующее проведения высокотехнологичных методов лечения;
- Необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для неотложной/экстренной госпитализации:
- Острые заболевания;
- Обострения хронических болезней;
- Отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии и другие угрожающие жизни острые состояния), а также круглосуточного медицинского наблюдении и проведения специальных видов обследования и лечения.
7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Препараты, которых следует избегать:
Барбитураты и их производные. Как правило, дети с инфантильными приступами лечатся фенобарбиталом**, наиболее часто используемым противоэпилептическим препаратом в этой возрастной группе. Исследования in vitro показывают, что барбитураты и их производные усугубляют транспортный дефект Glut1 в эритроцитах индивидуумов с синдромом дефицита Glut1 [36]. Иногда родители сообщали, что фенобарбитал** не улучшал контроль над приступами у ребенка или, возможно, ухудшал клиническое состояние ребенка.
Производные ксантина (например, кофеин), которые, как известно, ингибируют транспорт глюкозы с помощью Glut1 [86], также, как сообщается, ухудшают клиническое состояние пациентов с синдромом дефицита Glut1 [87]. Таким образом, пациентам рекомендуется избегать кофе и других напитков с кофеином.
|
N
|
Критерии качества
|
Оценка выполнения
|
|
1
|
Выполнено исследование уровня глюкозы в спинномозговой жидкости с параллельным исследованием уровня глюкозы в крови (при установлении диагноза)
|
Да/нет
|
|
2
|
Выполнено определение вариантов генов в образце биологического материала другом или неуточненном неклассифицированных в других рубриках методом секвенирования по Сенгеру и/или методом множественной лиганд-зависимой амплификации зондов (при установлении диагноза)
|
Да/нет
|
|
3
|
Выполнен прием (осмотр, консультация) врача-генетика первичный (при установлении диагноза)
|
Да/нет
|
|
4
|
Выполнен прием (осмотр, консультация) врача-невролога первичный и повторный
|
Да/нет
|
|
5
|
Выполнена электроэнцефалография
|
Да/нет
|
|
7
|
Выполнен прием (осмотр, консультация) врача-педиатра/врача-терапевта/врача общей практики первичный
|
Да/нет
|
Критерии оценки качества медицинской помощи
Список литературы
1. Coppola G., Veggiotti P., Cusmai R. et al. The ketogenic diet in children, adolescents and young adults with refractory epilepsy: an Italian multicentric experience. Epilepsy Res. 2002; 48(3): 221 - 227. doi: 10.1016/s0920-1211(01)00315-1
2. Усачева Е.Л., Айвазян С.О., Сорвачева Т.Н., Пырьева Е.А., Шорина М.Ю. Применение кетогенной диеты в лечении фармакорезистентных эпилепсий. Журнал неврологии и психиатрии им. С.С. Корсакова. 2004; 7: 29 - 34.
3. Сорвачева Т.Н., Пырьева Е.А., Конь И.Я., Усачева Е.Л., Айвазян С.О., Шорина М.Ю. Кетогенная диета в комплексном лечении фармакорезистентных форм эпилепсии у детей: клиническая оценка эффективности. Педиатрия. 2003; 2: 41 - 45.
4. Stafstrom C.E. Dietary approaches to epilepsy treatment: old and new options on the menu. Epilepsy Curr. 2004; 4(6): 215 - 222. doi: 10.1111/j.1535-7597.2004. 46001.x
5. National Library of Medicine, Genetics Home Reference your Guide to Understanding Genetic Conditions, GLUT1 deficiency syndrome. 2014.
6. Cheng C.M., Hicks K., Wang J., Eagles D.A., Bondy C.A. Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J Neurosci Res. 2004; 77(2): 270 - 276. doi: 10.1002/jnr.20144
7. Coman D.J., Sinclair K.G., Burke C.J., Appleton D.B. et al. Seizures, ataxia, developmental delay and the general paediatrician: glucose transporter 1 deficiency syndrome. J Paediatr Child Health. 2006 May; 42(5): 263 - 7. doi: 10.1111/j.1440-1754.2006.00852.x.
8. Larsen J., Johannesen K.M., Ek J., Tang S. et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015 Dec; 56(12): e203-8. doi: 10.1111/epi.13222.
9. Ramm-Pettersen A, Nakken KO, Haavardsholm KC, Selmer KK. GLUT1-deficiency syndrome: Report of a four-generation Norwegian family with a mild phenotype. Epilepsy Behav. 2017; 70 (Pt A): 1 - 4. doi: 10.1016/j.yebeh.2017.02.016
10. Ivanova N, Peycheva V, Kamenarova K, et al. Three novel SLC2A1 mutations in Bulgarian patients with different forms of genetic generalized epilepsy reflecting the clinical and genetic diversity of GLUT1-deficiency syndrome. Seizure. 2018; 54: 41 - 44. doi: 10.1016/j.seizure.2017.11.014
11. Liu Y, Bao X, Wang D, et al. Allelic variations of glut-1 deficiency syndrome: the chinese experience. Pediatr Neurol. 2012; 47(1): 30 - 34. doi: 10.1016/j.pediatrneurol.2012.04.010
12. Weber Y.G., Kamm C., Suls A. et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology. 2011; 77(10): 959 - 964. doi: 10.1212/WNL.0b013e31822e0479
13. Klepper J., Voit T. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain-- a review. Eur J Pediatr. 2002; 161(6): 295 - 304. doi: 10.1007/s00431-002-0939-3
14. Morris A.A. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005; 28(2): 109 - 121. doi: 10.1007/s10545-005-5518-0
15. Chinnery P.F. Defining neurogenetic phenotypes (or how to compare needles in haystacks). Brain. 2010; 133 (Pt3): 649 - 651. doi: 10.1093/brain/awq027
16. Leen W.G., Klepper J., Verbeek M.M. et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010; 133 (Pt 3): 655 - 670. doi: 10.1093/brain/awp336
17. Yang H., Wang D., Engelstad K. et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol. 2011; 70(6): 996 - 1005. doi: 10.1002/ana.22640
18. Wang D., Pascual J.M., De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews(R). Seattle (WA): University of Washington, Seattle; July 30, 2002 (last update March 3, 2018)
19. Лукьянова Е.Г., Айвазян С.О., Осипова К.В., Пырьева Е.А., Сорвачева Т.Н. Опыт применения кетогенной диеты у пациентов с синдромом дефицита транспортера глюкозы I типа (клиническое наблюдение)/Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2015. - Т. 115(5 - 2). - С. 53 - 60. - doi: 10.17116/jnevro20151155253-60.
20. Zorzi G., Castellotti B., Zibordi F., Gellera C., Nardocci N. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology. 2008; 71(2): 146 - 148. doi: 10.1212/01.wnl.0000316804.10020.ba
21. Urbizu A.,  E., Raspall-Chaure M. et al. Paroxysmal exercise-induced dyskinesia, writer"s cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J Neurol Sci. 2010; 295(1 - 2): 110 - 113. doi: 10.1016/j.jns.2010.05.017
E., Raspall-Chaure M. et al. Paroxysmal exercise-induced dyskinesia, writer"s cramp, migraine with aura and absence epilepsy in twin brothers with a novel SLC2A1 missense mutation. J Neurol Sci. 2010; 295(1 - 2): 110 - 113. doi: 10.1016/j.jns.2010.05.017
22. De Vivo D.C., Wang D. Glut1 deficiency: CSF glucose. How low is too low?. Rev Neurol (Paris). 2008; 164(11): 877 - 880. doi: 10.1016/j.neurol.2008.10.001
23. Overweg-Plandsoen W.C., Groener J.E., Wang D. et al. GLUT-1 deficiency without epilepsy--an exceptional case. J Inherit Metab Dis. 2003; 26(6): 559 - 563. doi: 10.1023/a: 1025999914822
24. Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, Shimizu T, Yudkoff M, Daikhin Y, Nissim I, Lazarow A. Brain amino acid metabolism and ketosis. The Journal of Neuroscience Journal of Neuroscience Research. 2001; 66: 272 - 281.
25. Leen W.G., Wevers R.A., Kamsteeg E.J., Scheffer H,. Verbeek MM, Willemsen MA. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol. 2013; 70(11): 1440 - 1444. doi: 10.1001/jamaneurol.2013.3090
26. Klepper J, Akman C, Armeno M, et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open. 2020; 5(3): 354 - 365. Published 2020 Aug 13. doi: 10.1002/epi4.12414
27. Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia. 2012; 53(9): 1503 - 1510. doi: 10.1111/j.1528-1167.2012.03592.x
28. Wolking S, Becker F, Bast T, et al. Focal epilepsy in glucose transporter type 1 (Glut1) defects: case reports and a review of literature. J Neurol. 2014; 261(10): 1881 - 1886. doi: 10.1007/s00415-014-7433-5
29. Peeraer A, Damiano JA, Bellows ST, et al. Evaluation of GLUT1 variation in non-acquired focal epilepsy. Epilepsy Res. 2017; 133: 54 - 57. doi: 10.1016/j.eplepsyres.2017.04.007
30. Pearson T.S., Pons R., Engelstad K., Kane S.A., Goldberg M.E., De Vivo D.C. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology. 2017; 88(17): 1666 - 1673. doi: 10.1212/WNL.0000000000003867.
31. Leary L.D., Wang D., Nordli D.R. Jr, Engelstad K., De Vivo D.C. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia. 2003; 44(5): 701 - 707. doi: 10.1046/j.1528-1157.2003.05302.x
32. Vining E.P., Freeman J.M., Ballaban-Gil K. et al. A multicenter study of the efficacy of the ketogenic diet. Arch Neurol. 1998; 55(11): 1433 - 1437. doi: 10.1001/archneur.55.11.1433
33. Усачева Е.Л., Айвазян С.О., Сорвачева Т.Н., Пырьева Е.А., Конь И.Я., Притыко А.Г., Шорина М.Ю., Осипова К.В. Кетогенная диета в лечении фармакорезистентных эпилепсий. Лечащий врач. 2004; 5: 46 - 50.
34. Кетогенная диета в лечении фармакорезистентных эпилепсий: показания к применению, критерии эффективности, осложнения: Методические рекомендации N 7, Правительство Москвы, Департамент здравоохранения. М.; 2005; 20.
35. Striano P., Weber Y.G., Toliat M.R. et al. GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology. 2012; 78(8): 557 - 562. doi: 10.1212/WNL.0b013e318247ff54
36. Klepper J., Fischbarg J., Vera J.C., Wang D., De Vivo D.C. GLUT1-deficiency: barbiturates potentiate haploinsufficiency in vitro. Pediatr Res. 1999; 46(6): 677 - 683. doi: 10.1203/00006450-199912000-00006
37. Pons R., Collins A., Rotstein M., Engelstad K., De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010; 25(3): 275 - 281. doi: 10.1002/mds.22808
38. Pearson T.S., Akman C., Hinton V.J., Engelstad K., De Vivo D.C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. 2013; 13(4): 342. doi: 10.1007/s11910-013-0342-7
39. Alter A.S., Engelstad K., Hinton V.J. et al. Long-term clinical course of Glut1 deficiency syndrome. J Child Neurol. 2015; 30(2): 160 - 169. doi: 10.1177/0883073814531822
40. Hully M, Vuillaumier-Barrot S, Le Bizec C, et al. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet. 2015; 58(9): 443 - 454. doi: 10.1016/j.ejmg.2015.06.007
41. Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009; 31(7): 545 - 552. doi: 10.1016/j.braindev.2009.02.008
42. Ito Y, Takahashi S, Kagitani-Shimono K, et al. Nationwide survey of glucose transporter-1 deficiency syndrome (GLUT-1DS) in Japan. Brain Dev. 2015; 37(8): 780 - 789. doi: 10.1016/j.braindev.2014.11.006
43. Willemsen MA, Vissers LE, Verbeek MM, et al. Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome. Eur J Hum Genet. 2017; 25(6): 771 - 774. doi: 10.1038/ejhg.2017.45
44. Friedman JR, Thiele EA, Wang D, et al. Atypical GLUT1 deficiency with prominent movement disorder responsive to ketogenic diet. Mov Disord. 2006; 21(2): 241 - 245. doi: 10.1002/mds.20660
45. Ebrahimi-Fakhari D., Pearl P.L. Movement disorders and inherited metabolic disorders - recognition, understanding, improving outcomes. - Cambridge University Press. - 2020. - 442 p.
46. Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol. 2014; 261(3): 589 - 599. doi: 10.1007/s00415-014-7240-z
47.  B., Prior C., Ma Q. et al. Childhood chorea with cerebral hypotrophy: a treatable GLUT1 energy failure syndrome. Arch Neurol. 2009; 66(11): 1410 - 1414. doi: 10.1001/archneurol.2009.236
B., Prior C., Ma Q. et al. Childhood chorea with cerebral hypotrophy: a treatable GLUT1 energy failure syndrome. Arch Neurol. 2009; 66(11): 1410 - 1414. doi: 10.1001/archneurol.2009.236
48. Suls A., Dedeken P., Goffin K. et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008; 131 (Pt 7): 1831 - 1844. doi: 10.1093/brain/awn113
49. Weber Y.G., Storch A., Wuttke T.V. et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008; 118(6): 2157 - 2168. doi: 10.1172/JCI34438.
50. Айвазян С.О., Лукьянова Е.Г., Ширяев Ю.С. Немедикаментозные методы лечения фармакорезистентной эпилепсии у детей. Эпилепсия и пароксизмальные состояния. 2014; 6: 1: 34 - 43.
51. Pascual J.M., Wang D., Yang R., Shi L., Yang H., De Vivo D.C. Structural signatures and membrane helix 4 in GLUT1: inferences from human blood-brain glucose transport mutants. J Biol Chem. 2008; 283(24): 16732 - 16742. doi: 10.1074/jbc.M801403200
52. Wang D., Yang H., Shi L. et al. Functional studies of the T295M mutation causing Glut1 deficiency: glucose efflux preferentially affected by T295M. Pediatr Res. 2008; 64(5): 538 - 543. doi: 10.1203/PDR.0b013e318184d2b5
53. Klepper J., Scheffer H., Leiendecker B. et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005; 36(5): 302 - 308. doi: 10.1055/s-2005-872843
54. Wang D., Pascual J.M., Yang H. et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005; 57(1): 111 - 118. doi: 10.1002/ana.20331
55. De Vivo D.C., Bohan T.P., Coulter D.L. et al. L-carnitine supplementation in childhood epilepsy: current perspectives. Epilepsia. 1998; 39(11): 1216 - 1225. doi: 10.1111/j.1528-1157.1998.tb01315.x
56. De Vivo D.C., Trifiletti R.R., Jacobson R.I., Ronen G.M., Behmand R.A., Harik S.I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991; 325(10): 703 - 709. doi: 10.1056/NEJM199109053251006
57. Pike M. Opsoclonus-myoclonus syndrome. Handb Clin Neurol. 2013; 112: 1209 - 1211. doi: 10.1016/B978-0-444-52910-7.00042-8
58. Тутельян В.А., Батурин А.К., Гаппаров М.Б. и др. Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации. 2009; МР 2.3.1.2432-08. - 40 с.
59. Wang D., Ho Y-Y, Pascual J.M., Hinton V. et al. GLUT1 deficiency syndrome: R333W genotype and paternal mosaicism. Ann Neurol. 2001; 50: S124.
60. Klepper J., Scheffer H., Elsaid M.F., Kamsteeg E.J. et al. Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics. 2009; 40(5): 207 - 210. doi: 10.1055/s-0030-1248264
61. Rotstein M., Engelstad K., Yang H. et al. Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol. 2010; 68(6): 955 - 958. doi: 10.1002/ana.22088
62. Freeman J., Veggiotti P., Lanzi G., Tagliabue A., Perucca E. The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Research. 2006; 68: 145 - 180. doi: 10.1016/j.eplepsyres.2005.10.003.
63. Roubergue A., Apartis E., Mesnage V. et al. Dystonic tremor caused by mutation of the glucose transporter gene GLUT1. J Inherit Metab Dis. 2011; 34(2): 483 - 488. doi: 10.1007/s10545-010-9264-6
64. Pascual JM, Ronen GM. Glucose Transporter Type I Deficiency (G1D) at 25 (1990 - 2015): Presumptions, Facts, and the Lives of Persons With This Rare Disease. Pediatr Neurol. 2015; 53(5): 379 - 393. doi: 10.1016/j.pediatrneurol.2015.08.001
65. Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Айвазян С.О., Осипова К.В., Сушко Л.М., Лукьянова Е.Г., Притыко А.Г. Синдром дефицита транспортера глюкозы I типа (болезнь де Виво): клинические и генетические аспекты. Медицинская генетика. 2016; 15(7): 28 - 32. https://doi.org/10.1234/XXXX-XXXX-2016-7-28-32
66. Shibata T, Kobayashi K, Yoshinaga H, Ono H, Shinpo M, Kagitani-Shimono K. Another Case of Glucose Transporter 1 Deficiency Syndrome with Periventricular Calcification, Cataracts, Hemolysis, and Pseudohyperkalemia. Neuropediatrics. 2017; 48(5): 390 - 393. doi: 10.1055/s-0037-1603520
67. Lukyanova E.G., Sushko L.M., Ayvazyan S.O., Osipova K.V., Pyreva E.A., Sorvacheva T.N., Zhilina S.S., Kozhanova T.V., Mescheryakova T.I. Glucose Transporter Type 1 Deficiency Syndrome (GLUT1) and using Ketogenic Diet in Treatment of De Vivo Disease: A Case Reports. Glob J Intellect Dev Disabil 2(3) 2017: 71 - 77.
68. Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology. 2010; 75(5): 432 - 440. doi: 10.1212/WNL.0b013e3181eb58b4
69. Kossoff E.H., Zupec-Kania B.A., Auvin S. et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018; 3(2): 175 - 192. Published 2018 May 21. doi: 10.1002/epi4.12225
70. Van der Louw E, van den Hurk D, Neal E, et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur J Paediatr Neurol. 2016; 20(6): 798 - 809. doi: 10.1016/j.ejpn.2016.07.009
71. Klepper J., Leiendecker B., Kossoff E.H. Pocket guide to the ketogenic diet. SPS publications, 54 p
72. Zupec-Kania B., Vanatta L., Johnson M. Ketogenic diet therapies for neurological disorders pocket guide for medical professionals. The Charlie foundation for ketogenic therapies 2018.
73. Vaudano A.E., Olivotto S., Ruggieri A. et al. Brain correlates of spike and wave discharges in GLUT1 deficiency syndrome. Neuroimage Clin. 2016; 13: 446 - 454. Published 2016 Dec 21. doi: 10.1016/j.nicl.2016.12.026
74. Ismayilova N, Hacohen Y, MacKinnon AD, Elmslie F, Clarke A. GLUT-1 deficiency presenting with seizures and reversible leukoencephalopathy on MRI imaging. Eur J Paediatr Neurol. 2018; 22(6): 1161 - 1164. doi: 10.1016/j.ejpn.2018.02.002
75. Zeng W., Xing Z., Tan M. et al. Propofol regulates activated macrophages metabolism through inhibition of ROS-mediated GLUT1 expression. Inflamm. Res. 70, 473 - 481 (2021). doi: 10.1007/s00011-021-01449-y
76. Schiller K., Avigdor T., Kortas A., Kunz M. et al. Monitoring Glucose Concentrations in Children with Epilepsy on a Ketogenic Diet. Healthcare (Basel). 2022 Jan 27; 10(2): 245. doi: 10.3390/healthcare10020245
77. Ketogenic diet - managing complications, version 1. Cambridge University Hospitals NHS Foundation Trust, 12 p
78. Baumeister FA, Oberhoffer R, Liebhaber GM, et al. Fatal propofol infusion syndrome in association with ketogenic diet. Neuropediatrics. 2004; 35(4): 250 - 252. doi: 10.1055/s-2004-820992
79. Yudkoff M., Daikhin Y., Nissim I., Lazarow A., Nissim I. Brain amino acid metabolism and ketosis. J Neurosci Res. 2001; 66(2): 272 - 281. doi: 10.1002/jnr.1221
80. Akman C.I., Provenzano F., Wang D. et al. Topography of brain glucose hypometabolism and epileptic network in glucose transporter 1 deficiency. Epilepsy Res. 2015; 110: 206 - 215. doi: 10.1016/j.eplepsyres.2014.11.007
81. Ruiz Herrero J.,  Villarroya E.,
Villarroya E., 
 L. et al. Classic Ketogenic Diet and Modified Atkins Diet in SLC2A1 Positive and Negative Patients with Suspected GLUT1 Deficiency Syndrome: A Single Center Analysis of 18 Cases. Nutrients. 2021 Mar 4; 13(3): 840. doi: 10.3390/nu13030840.
L. et al. Classic Ketogenic Diet and Modified Atkins Diet in SLC2A1 Positive and Negative Patients with Suspected GLUT1 Deficiency Syndrome: A Single Center Analysis of 18 Cases. Nutrients. 2021 Mar 4; 13(3): 840. doi: 10.3390/nu13030840.
82. Butte NF, Watson KB, Ridley K, et al. A Youth Compendium of Physical Activities: Activity Codes and Metabolic Intensities. Med Sci Sports Exerc. 2018; 50(2): 246 - 256. doi: 10.1249/MSS.0000000000001430
83. Daci A, Bozalija A, Jashari F, Krasniqi S. Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. Int J Mol Sci. 2018; 19(1): 122. Published 2018 Jan 5. doi: 10.3390/ijms19010122
84. Adeva-Andany MM,  C,
C,  D, Castro-Quintela E,
D, Castro-Quintela E,  A. Sodium bicarbonate therapy in patients with metabolic acidosis. ScientificWorldJournal. 2014; 2014: 627673. doi: 10.1155/2014/627673
A. Sodium bicarbonate therapy in patients with metabolic acidosis. ScientificWorldJournal. 2014; 2014: 627673. doi: 10.1155/2014/627673
85. Gras D, Roze E, Caillet S,  A, Doummar D, Billette de Villemeur T, Vidailhet M, Mochel F. GLUT1 deficiency syndrome: an update. Rev Neurol (Paris). 2014 Feb; 170(2): 91 - 9. doi: 10.1016/j.neurol.2013.09.005.
A, Doummar D, Billette de Villemeur T, Vidailhet M, Mochel F. GLUT1 deficiency syndrome: an update. Rev Neurol (Paris). 2014 Feb; 170(2): 91 - 9. doi: 10.1016/j.neurol.2013.09.005.
86. Ho Y.Y., Yang H, Klepper J, Fischbarg J. et al. Glucose transporter type 1 deficiency syndrome (Glut1DS): methylxanthines potentiate GLUT1 haploinsufficiency in vitro. Pediatr Res. 2001; 50(2): 254 - 260. doi: 10.1203/00006450-200108000-00015
87. Brockmann K., Wang D., Korenke C.G. et al. Autosomal dominant glut-1 deficiency syndrome and familial epilepsy. Ann Neurol. 2001; 50(4): 476 - 485. doi: 10.1002/ana.1222
88. Liu YY, Bao XH, Wang S, Fu N, Liu XY, Song FY, Yang YL, Wu Y, Zhang YH, Wu JX, Jiang YW, Qin J, Wu XR. [Clinical and genetic characteristics of glucose transporter type 1 deficiency syndrome]. Zhonghua Er Ke Za Zhi. 2013 Jun; 51(6): 443 - 7. Chinese. PMID: 24120063.
89. Blau N., Hoffmann G.F., Leonard J., Clarke J.T.R. Physician's Guide to the Treatment and Follow-Up of Metabolic Diseases. - 2006 - Springer - 404 p.
90.  A, Roze E. GLUT1 Deficiency in a Patient Diagnosed as Cerebral Palsy: Is NGS a Valuable Tool to Be Considered in All Cases of CP to Detect Underlying Genetic Disorders? Mov Disord Clin Pract. 2019 Mar 28; 6(4): 277 - 279. doi: 10.1002/mdc3.12754.
A, Roze E. GLUT1 Deficiency in a Patient Diagnosed as Cerebral Palsy: Is NGS a Valuable Tool to Be Considered in All Cases of CP to Detect Underlying Genetic Disorders? Mov Disord Clin Pract. 2019 Mar 28; 6(4): 277 - 279. doi: 10.1002/mdc3.12754.
91. Berry-Kravis E., Booth G., Sanchez A.C., Woodbury-Kolb J. Carnitine levels and the ketogenic diet. - 2001 - Epilepsia - 42(11): 1445 - 1451
92. Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. 2002 Jul 30 [Updated 2018 Mar 1]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993 - 2023.
93. Кулиш Е.А., Котов А.С., Мухина Е.В. и др. Синдром дефицита транспортера глюкозы I типа: клинический случай. Русский журнал детской неврологии 2019; 14(2): 23 - 8
94. Ito Y, Oguni H, Ito S, Oguni M, Osawa M. A modified Atkins diet is promising as a treatment for glucose transporter type 1 deficiency syndrome. Dev Med Child Neurol. 2011; 53(7): 658 - 663. doi: 10.1111/j.1469-8749.2011.03961.x
95. Tzadok M, Nissenkorn A, Porper K, et al. The many faces of Glut1 deficiency syndrome. J Child Neurol. 2014; 29(3): 349 - 359. doi: 10.1177/0883073812471718
96. Лукьянова Е.Г., Айвазян С.О., Осипова К.В., Пырьева Е.А., Сорвачева Т.Н. Опыт применения кетогенной диеты у пациентов с синдромом дефицита транспортера глюкозы I типа (клиническое наблюдение). Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2015; 115(5 - 2): 53 - 60.
97. Zanaboni MP, Pasca L, Geraci MA, Varesio C, Guglielmetti M, Tagliabue A, Grumi S, De Giorgis V. Case report: KETOLAND the psychoeducation program for ketogenic diet. Front Psychiatry. 2023 Jun 8; 14: 1155717. doi: 10.3389/fpsyt.2023.1155717.
98. Winczewska-Wiktor A, Hoffman-Zacharska D, Starczewska M, Kaczmarek I, Badura-Stronka M, Steinborn B. Variety of symptoms of GLUT1 deficiency syndrome in three-generation family. Epilepsy Behav. 2020 May; 106: 107036. doi: 10.1016/j.yebeh.2020.107036. Epub 2020 Apr 1. PMID: 32247176.
99. Diaz-Arias LA, Henry-Barron BJ, Buchholz A, Cervenka MC. Positive impact of a modified Atkins diet on cognition, seizure control, and abnormal movements in an adult with glucose transporter type 1 deficiency syndrome: case report. Neurol Sci. 2022 May; 43(5): 3449 - 3452.
100. Hussain N., Khan A., Kearns J., Samanta R., Wilford E. Management of children on ketogenic diet for epilepsy. 12.2022.
101. Kloka J., Kranepuhl S., Zacharowski K., Raimann F.J. Total Intravenous Anesthesia in GLUT1 Deficiency Syndrome Patient: A Case Report. Am J Case Rep. 2019 May 5; 20: 647 - 650.
102. Van S, Lam V, Patel K, Humphries A, Siddiqi J. Propofol-Related Infusion Syndrome: A Bibliometric Analysis of the 100 Most-Cited Articles. Cureus. 2023 Oct 4; 15(10): e46497
103. Singh A, Anjankar AP. Propofol-Related Infusion Syndrome: A Clinical Review. Cureus. 2022 Oct 17; 14(10): e 30383
104. https://grls.minzdrav.gov.ru/
105. Kolic I, Radic Nisevic J, Vlasic Cicvaric I, Butorac Ahel I, Lah Tomulic K, Segulja S, Baraba Dekanic K, Serifi S, Ovuka A, Prpic I. GLUT1 Deficiency Syndrome-Early Treatment Maintains Cognitive Development? (Literature Review and Case Report). Genes (Basel). 2021 Aug 31; 12(9): 1379
106. Phillips, Matthew. (2019). Ketogenic Diet Therapies in Children and Adults with Epilepsy. 10.5772/intechopen.83711.
107. Belousova ED. Snizhenie kontsentratsii karnitina u patsientov s  [The decreased level of plasma carnitine in patients with epilepsy]. Zh Nevrol Psikhiatr Im S S Korsakova. 2017; 117(6): 106 - 110. Russian. doi: 10.17116/jnevro201711761106-110. PMID: 28745680.
[The decreased level of plasma carnitine in patients with epilepsy]. Zh Nevrol Psikhiatr Im S S Korsakova. 2017; 117(6): 106 - 110. Russian. doi: 10.17116/jnevro201711761106-110. PMID: 28745680.
Приложение А1
СОСТАВ
РАБОЧЕЙ ГРУППЫ ПО РАЗРАБОТКЕ И ПЕРЕСМОТРУ
КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
1. Айвазян Сергей Оганесович - к.м.н., ведущий научный сотрудник, доцент, научный руководитель группы резистентных форм эпилепсии научного отдела ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы; Медицинский центр "Невромед", Москва
2. Анисимова Инга Вадимовна - к.м.н., заведующая отделом организации медицинской помощи - врач-генетик ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Ассоциации медицинских генетиков
3. Баранов Александр Александрович - акад. РАН, профессор, д.м.н.; почетный президент Союза педиатров России, советник руководителя НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", профессор кафедры педиатрии и детской ревматологии ФГАОУ "Первый МГМУ им. И.М. Сеченова" Минздрава России (Сеченовский Университет), главный внештатный специалист педиатр Минздрава России
4. Белоусова Елена Дмитриевна - д.м.н., профессор, заслуженный врач РФ, руководитель отдела психоневрологии и эпилептологии Обособленного структурного подразделения "Научно-исследовательского клинического института педиатрии имени академика Ю.А. Вельтищева" ФГАОУ ВО МЗ РФ "РНИМУ им. Н.И. Пирогова"
5. Вашакмадзе Нато Джумберовна - д.м.н., руководитель отдела орфанных болезней и профилактики инвалидизирующих заболеваний НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", профессор кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, член Союза педиатров России
6. Вишнева Елена Александровна - д.м.н., профессор РАН, заместитель руководителя НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", Минобрнауки по научной работе, профессор кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, член Союза педиатров России
7. Жилина Светлана Сергеевна - к.м.н., доцент, ведущий научный сотрудник генетической группы научного отдела, врач-генетик ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы; ФГАОУ ВО "Российский национальный исследовательский медицинский университет имени Н.И. Пирогова" МЗ РФ
8. Журкова Наталия Вячеславовна - к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", член Союза педиатров России, член Ассоциации медицинских генетиков
9. Зарубина Вера Владимировна - врач-генетик медико-генетического отделения Морозовской ДГКБ ДЗМ
10. Захарова Екатерина Юрьевна - д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков, член Европейского общества по изучению наследственных болезней обмена веществ (SSIEM)
11. Кожанова Татьяна Викторовна - к.м.н., доцент, ведущий научный сотрудник генетической группы научного отдела, врач-лабораторный генетик ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы; ФГАОУ ВО "Российский национальный исследовательский медицинский университет имени Н.И. Пирогова" МЗ РФ
12. Кузенкова Людмила Михайловна - д.м.н., заведующая отделением психоневрологии и психосоматической патологии ФГАУ "Научный медицинский исследовательский центр здоровья детей" МЗ РФ, член Союза педиатров России
13. Куцев Сергей Иванович - акад. РАН, д.м.н., директор ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", Президент Ассоциации медицинских генетиков (АМГ)
14. Лукьянова Екатерина Геннадьевна - врач-невролог ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы
15. Михайлова Светлана Витальевна - д.м.н., заведующая отделением ФГБУ "Российская Детская Клиническая Больница" МЗ РФ
16. Намазова-Баранова Лейла Сеймуровна - акад. РАН, профессор, д.м.н., президент Союза педиатров России; паст-президент EPA/UNEPSA; руководитель НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", заведующая кафедрой факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, главный внештатный детский специалист по профилактической медицине Минздрава России
17. Никитин Сергей Сергеевич - д.м.н., профессор, председатель "Общества специалистов по нервно-мышечным заболеваниям"
18. Осипова Карина Вартановна - к.м.н., заведующая психоневрологическим отделением N 1 ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы; Медицинский центр "Невромед", Москва
19. Подклетнова Татьяна Владимировна - к.м.н., с.н.с. лаборатории редких наследственных болезней у детей ФГАУ "Научный медицинский исследовательский центр здоровья детей" МЗ РФ, член Союза педиатров России
20. Пырьева Екатерина Анатольевна - к.м.н., нутрициолог, заведующий лабораторией возрастной нутрициологии ФГБУН "ФИЦ питания, биотехнологии и безопасности пищи"
21. Репина Светлана Афанасьевна - к.м.н., врач-генетик ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков, член Ассоциации медицинских генетиков
22. Свиридова Валерия Валерьевна - врач-генетик отдела организации медицинской помощи ФГБНУ "МГНЦ", м.н.с. лаборатории мутагенеза ФГБНУ "МГНЦ"
23. Селимзянова Лилия Робертовна - к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", доцент кафедры педиатрии и детской ревматологии ФГАОУ "Первый МГМУ им. И.М. Сеченова" Минздрава России (Сеченовский Университет), доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, член Союза педиатров России
24. Смирнова Ольга Яковлевна - врач-генетик, старший научный сотрудник отдела стандартизации и изучения основ доказательной медицины НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", член Союза педиатров России
25. Сурков Андрей Николаевич, д.м.н., заведующий отделением гастроэнтерологии для детей стационара для детей, заведующий отделом научных основ детской гастроэнтерологии, гепатологии и метаболических нарушений НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", профессор кафедры факультетской педиатрии педиатрического факультета ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
26. Сорвачева Татьяна Николаевна - д.м.н., заведующая кафедрой диетологии и нутрициологии ФГБОУ ДПО "Российская медицинская академия непрерывного последипломного образования"
27. Сушко Лилия Марленовна - врач-невролог ГБУЗ "Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого" ДЗ г. Москвы
28. Субботин Дмитрий Михайлович - врач-генетик ФГБНУ "МГНЦ", член Ассоциации медицинских генетиков
29. Тепаев Рустэм Фаридович, д.м.н., профессор кафедры педиатрии с курсом детской ревматологии ФГАОУ "Первый МГМУ им. И.М. Сеченова" Минздрава России (Сеченовский Университет), руководитель службы анестезиологии-реанимации ДГКБ им З.А. Башляевой ДЗМ
30. Федосеенко Марина Владиславовна, к.м.н., заведующая отделом разработки научных подходов к иммунизации пациентов с отклонениями в состоянии здоровья и хроническими болезнями, ведущий научный сотрудник, врач-педиатр НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им. акад. Б.В. Петровского", доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, член Союза педиатров России
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2
МЕТОДОЛОГИЯ РАЗРАБОТКИ КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
Целевая аудитория данных клинических рекомендаций:
1. Врачи-генетики;
2. Врачи-диетологи;
3. Врачи - лабораторные генетики;
4. Врачи-неврологи;
5. Врачи общей практики (семейные врачи);
6. Врачи-педиатры;
7. Врачи-терапевты;
8. Врачи функциональной диагностики;
9. Врачи-психиатры;
10. Врачи-психиатры детские;
11. Медицинские психологи;
12. Обучающиеся в ординатуре;
13. Студенты медицинских ВУЗов.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
|
УУДД
|
Расшифровка
|
|
1
|
Систематические обзоры исследований с контролем референсным методом или систематический обзор рандомизированных клинических исследований с применением метаанализа
|
|
2
|
Отдельные исследования с контролем референсным методом или отдельные рандомизированные клинические исследования и систематические обзоры исследований любого дизайна, за исключением рандомизированных клинических исследований, с применением метаанализа
|
|
3
|
Исследования без последовательного контроля референсным методом или исследования с референсным методом, не являющимся независимым от исследуемого метода или нерандомизированные сравнительные исследования, в том числе когортные исследования
|
|
4
|
Несравнительные исследования, описание клинического случая
|
|
5
|
Имеется лишь обоснование механизма действия или мнение экспертов
|
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
|
УУДД
|
Расшифровка
|
|
1
|
Систематический обзор РКИ с применением метаанализа
|
|
2
|
Отдельные РКИ и систематические обзоры исследований любого дизайна, за исключением РКИ, с применением метаанализа
|
|
3
|
Нерандомизированные сравнительные исследования, в т.ч. когортные исследования
|
|
4
|
Несравнительные исследования, описание клинического случая или серии случаев, исследования "случай-контроль"
|
|
5
|
Имеется лишь обоснование механизма действия вмешательства (доклинические исследования) или мнение экспертов
|
Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
|
УУР
|
Расшифровка
|
|
A
|
Сильная рекомендация (все рассматриваемые критерии эффективности (исходы) являются важными, все исследования имеют высокое или удовлетворительное методологическое качество, их выводы по интересующим исходам являются согласованными)
|
|
B
|
Условная рекомендация (не все рассматриваемые критерии эффективности (исходы) являются важными, не все исследования имеют высокое или удовлетворительное методологическое качество и/или их выводы по интересующим исходам не являются согласованными)
|
|
C
|
Слабая рекомендация (отсутствие доказательств надлежащего качества (все рассматриваемые критерии эффективности (исходы) являются неважными, все исследования имеют низкое методологическое качество и их выводы по интересующим исходам не являются согласованными)
|
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию - не реже, чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утвержденным КР, но не чаще 1 раза в 6 месяцев.
Приложение А3
СПРАВОЧНЫЕ МАТЕРИАЛЫ,
ВКЛЮЧАЯ СООТВЕТСТВИЕ ПОКАЗАНИЙ К ПРИМЕНЕНИЮ
И ПРОТИВОПОКАЗАНИЙ, СПОСОБОВ ПРИМЕНЕНИЯ И ДОЗ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ, ИНСТРУКЦИИ ПО ПРИМЕНЕНИЮ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА
1. Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" (N 323-ФЗ от 21.11.2011).
2. Постановление Правительства РФ от 30 июля 1994 г. N 890 "О государственной поддержке развития медицинской промышленности и улучшении обеспечения населения и учреждений здравоохранения лекарственными средствами и изделиями медицинского назначения" (с изменениями и дополнениями).
3. Приказ Министерства здравоохранения РФ "Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями" от 21.04.2022 N 274н.
4. Постановление Правительства N 403 от 26.04.2012 г. "О порядке ведения Федерального регистра лиц, страдающих угрожающими и хронически прогрессирующими редкими орфанными заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности и его регионального сегмента".
5. Приказ Министерства здравоохранения Российской Федерации от 15 декабря 2014 г. N 834н "Об утверждении унифицированных форм медицинской документации, используемых в медицинских организациях, оказывающих медицинскую помощь в амбулаторных условиях, и порядков по их заполнению".
6. Приказ Министерства здравоохранения Российской Федерации от 20 декабря 2012 г. N 1177н "Об утверждении порядка дачи информированного добровольного согласия на медицинское вмешательство и отказа от медицинского вмешательства в отношении определенных видов медицинских вмешательств, форм информированного добровольного согласия на медицинское вмешательство и форм отказа от медицинского вмешательства".
7. Приказ Минздрава России (Министерство здравоохранения РФ) от 24 ноября 2021 г. N 1094н "Об утверждении Порядка назначения лекарственных препаратов, форм рецептурных бланков на лекарственные препараты, Порядка оформления указанных бланков, их учета и хранения, форм бланков рецептов, содержащих назначение наркотических средств или психотропных веществ, Порядка их изготовления, распределения, регистрации, учета и хранения, а также Правил оформления бланков рецептов, в том числе в форме электронных документов"
8. Приказ Министерства здравоохранения Российской Федерации от 16 мая 2019 г. N 302н "Об утверждении Порядка прохождения несовершеннолетними диспансерного наблюдения, в том числе в период обучения и воспитания в образовательных организациях"
9. Распоряжение Правительства РФ от 31 декабря 2018 г. N 3053-р "Об утверждении перечней медицинских изделий, имплантируемых в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи и отпускаемых по рецептам на медицинские изделия при предоставлении набора социальных услуг"
10. Приказ Министерства здравоохранения РФ от 6 июня 2013 г. N 354н "О порядке проведения патологоанатомических вскрытий" (зарегистрирован Министерством юстиции Российской Федерации 16 декабря 2013 г., регистрационный N 30612).
Информация о лекарственных средствах: https://grls.rosminzdrav.ru/
Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи:
1. Федеральный закон "О внесении изменений в Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам оказания паллиативной медицинской помощи" от 06.03.2019 N 18-ФЗ.
2. Приказ Минздрава России N 345н, Минтруда России от 31.05.2019 N 372н "Об утверждении положения об организации оказания паллиативной медицинской помощи, включая порядок взаимодействия медицинских организаций, организаций социального обслуживания и общественных объединений, иных некоммерческих организаций, осуществляющих свою деятельность в сфере охраны здоровья".
3. Приказ Минздрава России N 348н от 31 мая 2019 года "Об утверждении перечня медицинских изделий, предназначенных для поддержания органов и систем организма человека, предоставляемых для использования на дому".
4. Приказ Минздрава России N 505н от 10 июля 2019 года "Об утверждении Порядка передачи от медицинской организации пациенту (его законному представителю) медицинских изделий, предназначенных для поддержания функций органов и систем организма человека, для использования на дому при оказании паллиативной медицинской помощи".
5. Приказ МЗ РФ N 831 от 3 октября 2019 года "Об утверждении ведомственной целевой программы "Развитие системы оказания паллиативной медицинской помощи".
Прочие нормативно-правовые документы, с учетом которых разработаны клинические рекомендации:
6. Международная классификация болезней, травм и состояний, влияющих на здоровье (МКБ-10);
7. Приказ МЗ РФ от 20 декабря 2012 г. N 1183н "Об утверждении номенклатуры должностей медицинских работников и фармацевтических работников".
8. Приказ МЗ РФ от 23 июля 2010 г. N 541н. Единый квалификационный справочник должностей руководителей, специалистов и служащих, раздел Квалификационные характеристики должностей работников в сфере здравоохранения.
9. Федеральный закон от 25.12.2018 N 489 489-ФЗ "О внесении изменений в статью 40 Федерального закона "Об обязательном медицинском страховании в Российской Федерации" и Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам клинических рекомендаций".
10. Приказ Минздрава России N 103н от 28.02.2019 г. "Об утверждении порядка и сроков разработки клинических рекомендаций, их пересмотра, типовой формы клинических рекомендаций и требований к их структуре, составу и научной обоснованности, включаемой в клинические рекомендации информации".
11. Приказ Минздрава России от 13.10.2017 N 804н "Об утверждении номенклатуры медицинских услуг".
12. Приказ Министерства труда и социальной защиты РФ от 27 августа 2019 г. N 585н "О классификациях и критериях, используемых при осуществлении медико-социальной экспертизы граждан федеральными государственными учреждениями медико-социальной экспертизы";
13. Приказ Министерства здравоохранения и социального развития Российской Федерации "О порядке применения лекарственных средств у больных по жизненным показаниям" от 9 августа 2005 г. N 494
14. Информационное письмо Минздрава России по возможности закупки лекарственного препарата по торговому наименованию (https://www.rosminzdrav.ru/news/2019/12/18/13043-minzdrav-podgotovil-informatsionnoe-pismo-po-vozmozhnosti-zakupki-lekarstvennogo-preparata-po-torgovomu-naimenovaniyu);
15. Перечень специализированных продуктов лечебного питания для детей-инвалидов на 2023 год, Распоряжение Правительства Российской Федерации от 11 декабря 2023 г. N 3551-р
Приложение Б
АЛГОРИТМЫ ДЕЙСТВИЙ ВРАЧА
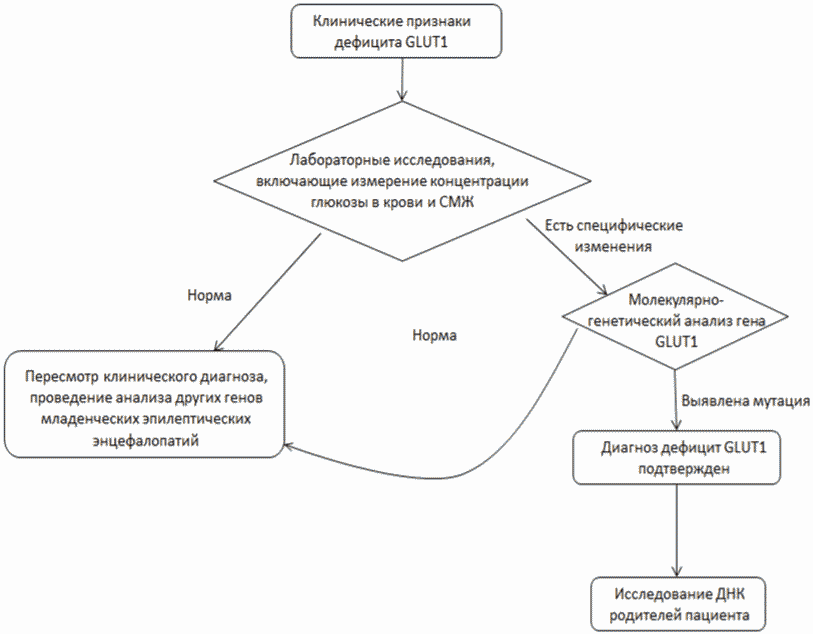
Приложение В
ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Что такое синдром дефицита Glut1?
Синдром дефицита Glut1 (синдром недостаточности транспортера глюкозы I типа) - это редкое генетическое заболевание с преимущественным поражением центральной нервной системы. Причиной заболевания является дефект в гене SLC2A1, который отвечает за работу белка-транспортера глюкозы 1 типа (Glut1), отвечающего за поступление глюкозы из крови в головной мозг. В результате генетического дефекта функция белка нарушается и головной мозг лишается основного источника энергии. У ребенка более 85% всей глюкозы расходуется на нужды именно нервной системы. В крови при этом сохраняется нормальный уровень глюкозы, но нарушение работы переносчика глюкозы в центральную нервную систему приводит к тому, что снижается концентрация глюкозы в ликворе. Это приводит к голоданию клеток головного мозга, а значит, к развитию неврологической симптоматики - судорогам, нарушениям движений, речи и другим проявлениям. Заболевание впервые описано Де Виво в 1991 году.
Как проявляется синдром дефицита Glut1?
Для заболевания характерно развитие метаболической энцефалопатии, сопровождающейся эпилептическими приступами, задержкой психомоторного развития, нарушением координации (атаксией), изменением мышечного тонуса (дистонией), приобретенной микроцефалией (уменьшением темпов увеличения окружности головы) и другими неврологическими проявлениями. Однако клинические проявления заболевания вариабельны и могут отличаться даже в пределах одной семьи.
Как устанавливается диагноз синдром дефицита Glut1?
Диагноз заболевания устанавливается на основании клинической картины и результатов лабораторных исследований. Диагностическим критерием заболевания служит снижение глюкозы в спинномозговой жидкости ниже порогового уровня на фоне нормального уровня глюкозы в крови. Окончательный диагноз устанавливается после генетического обследования - ДНК-диагностики.
Как наследуется синдром дефицита Glut1?
Заболевание наследуется по аутосомно-доминантному типу (рис. В1.1), то есть одной мутации достаточно, чтобы заболевание проявилось. У большинства пациентов с синдромом дефицита Glut1 мутации возникают de novo, т.е. впервые (не наследуются от родителей). Реже заболевание может передаться от пораженного родителя, носителя патогенной мутации, имеющего более легкую форму заболевания. Риск передачи поврежденного гена от пораженного родителя потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Аутосомно-доминантный тип наследования
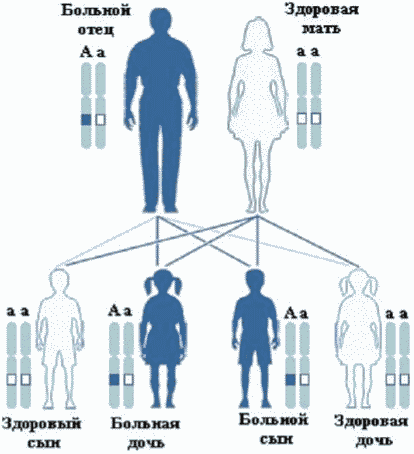
Рисунок В1.1 Аутосомно-доминантный тип наследования.
Как лечат пациентов с синдромом дефицита Glut1?
В настоящее время единственным эффективным методом терапии пациентов с синдромом дефицита Glut1 является кетогенная диета - высокожировая, низкоуглеводная диета.
Терапевтическое действие КД заключается в том, что наш мозг может использовать в качестве энергии не только глюкозу, но и кетоновые тела (3-гидроксибутират, ацетоацетат). Это водорастворимые соединения, синтезируемые в клетках печени из жирных кислот (а именно из них состоят жиры) при недостаточном количестве углеводов. Кетоновые тела способны проникать в головной мозг посредством другого белка-транспортера, обеспечивая мозг необходимым количеством энергии для метаболизма, и тем самым предотвращая дальнейшее его повреждение и способствуя уменьшению клинических проявлений заболевания, купированию судорог, улучшению двигательных, речевых и когнитивных функций.
Чтобы постоянно поддерживать необходимую концентрацию кетонов в крови, ежедневный рацион пациента обогащается жирами. А чтобы метаболизм сдвигался в сторону расщепления жиров и получения энергии за счет кетоновых тел, ограничивается поступление углеводов.
Диета имеет некоторые побочные эффекты и особенности, но при правильном подходе и расчетах хорошо переносится. Введение в кетоз осуществляется строго под медицинским наблюдением. Врач поможет подобрать нужное соотношение жиров в рационе под контролем уровня кетонов в крови и контролем функций других органов.
Прогноз заболевания синдром дефицита Glut1
Прогноз зависит от типа заболевания, возраста манифестации и начала терапии. При ранней диагностики и своевременной терапии, прогноз, как правило, благоприятный, пациенты могут рассчитывать на полноценную жизнь, отличающуюся лишь образом жизни и питанием.
Где в России занимаются диагностикой и лечением синдрома дефицита Glut1?
В России есть много центров, которые проводят молекулярно-генетические диагностические тесты. В каждом из регионов существуют медико-генетические центры, куда пациенты могут обратиться за консультацией врача-генетика и назначением молекулярных исследований. Кетогенную диету назначает врач-невролог, врач-диетолог, врач-гастроэнтеролог, врач-педиатр со знаниями диетологии.
Узнать больше
В мире есть много организаций, которые могут помочь вам найти ответы на любые другие вопросы, связанных с синдромом дефицита Glut1:
Международное объединение семей с дефицитом Glut1 https://www.g1dfoundation.org
Российский портал об эпилепсиях https://epilepsyinfo.ru
Международный портал по редким болезням www.orpha.net
Европейская организация, объединяющая пациентов с разными редкими заболеваниями EURORDIS www.eurordis.org
Приложение В2
ПРИМЕР
ЭКСТРЕННОЙ ПАМЯТКИ ДЛЯ ПАЦИЕНТОВ, НАХОДЯЩИХСЯ
НА КЕТОГЕННОЙ ДИЕТЕ
Внимание! Пациент находится на кетогенной диете.
Кетогенная диета (КД) - диета под медицинским наблюдением, высокожировая, низкоуглеводная. Является одним из способов лечения некоторых фармакорезистентных эпилепсий. Она индивидуально подбирается для каждого ребенка, поэтому предписания по калорийности и соотношению белков, жиров и углеводов специфичны для каждого ребенка.
При соблюдении кетогенной диеты целевые значения кетонов:
В КРОВИ - 2 - 5 ММОЛЬ/Л (допустимое значение до 6 при хорошем самочувствии),
В МОЧЕ - 8 - 16 ММОЛЬ/Л.
При снижении уровня кетонов в крови возможно возвращение/усиление приступов.
Диета имеет некоторые побочные эффекты и особенности, но обычно неплохо переносится. Многие виды недомогания могут лечиться амбулаторно. Пациент на КД обычно нуждается в стационарном наблюдении, если имеет следующие жалобы:
- Рвота и диарея (обычно вызваны гастроэнтеритом)
- Фебрильная лихорадка
- Учащение эпилептических приступов.
ПОМОЩЬ:
4 ключевых фактора при обращении за медицинской помощью:
1. Определение причины недомогания (при признаках сепсиса, рвоты с кровью или желчью необходима госпитализация в стационар)
2. Определение уровня обезвоживания
3. Выявление кетоацидоза (определение кислотно-щелочного состояния и газов крови крови (исследование уровня водородных ионов (pH) крови, исследование уровня буферных веществ в крови), уровня глюкозы в крови, исследование уровня натрия в крови, уровня общего кальция в крови, уровня калия в крови, уровня хлоридов в крови, исследование уровня молочной кислоты в крови; слабость, тошнота, фруктовый (кетоновый) запах в выдыхаемом воздухе, тахикардия, тахипноэ)
4. Определение отклонения от обычного уровня кетонемии - пациенты и их родители должны знать обычный уровень кетонемии при хорошем самочувствии ребенка:
- При кетонемии более 5 ммоль/л и отсутствии клинических признаков кетоза необходимо наблюдение,
- Кетонемия ниже 2 ммоль/л НЕ является опасной, но нежелательна.
ОКАЗАНИЕ ПОМОЩИ:
I. Обезвоживание
1. При легком обезвоживании:
Оральная регидратация:
Увеличение количества обычных напитков - вода, чай без сахара.
Нельзя! Употребление напитков с сахаром, соков, молока.
Кето-жидкости разрешены.
Если пациент не пьет - контролируйте уровень обезвоживания, измените лечение при необходимости.
2. Умеренное обезвоживание, но ребенок охотно пьет, нет рвоты
Оральная регидратация, могут использоваться регидрататирующие солевые препараты для перорального приема - Декстроза + Калия хлорид + Натрия хлорид + Натрия цитрат** - содержат, соответственно, 1 и 1,5 г декстрозы** на 100 мл раствора
Регидратация более важна, чем снижение целевого уровня кетоза!
3. Умеренное обезвоживание, но ребенок не хочет пить
При отсутствии рвоты возможна регидратация через назогастральный зонд - Декстроза + Калия хлорид + Натрия хлорид + Натрия цитрат** + (Кето-жидкости при отсутствии гиперкетоза).
4. Выраженное обезвоживание
Любой ребенок с тяжелым обезвоживанием нуждается во внутривенном введении 0,9% раствора натрия хлорида** 10 - 20 мл/кг, далее при необходимости могут быть введены другие растворы.
НЕТ необходимости во введении раствора декстрозы** при гликемии более 2,6 ммоль/л.
II. Гипогликемия
Уровень глюкозы ниже 2,6 ммоль/л:
Проверьте уровень кетонов. Если они в нужных значениях, а пациент неврологически стабильный - допустите гипогликемию. Если возникают неврологические симптомы - пероральный прием углеводов.
2 - 5 г углеводов:
- 20 - 50 мл фруктового сока
- 1 чайная ложка сахара, варенье, мед или сироп (можно с водой, чаем)
- 5 г (1 чайная ложка) #декстрозы** порошок в 100 мл воды [100]
- Можно использовать 10% раствор декстрозы** (10 мл - 1 г декстрозы**)
Уровень глюкозы выше 2,6 ммоль/л, но есть симптомы гипогликемии:
Ребенок вялый, снижен тонус, бледный, летаргия, снижена температура тела, холодный и липкий пот, цианоз. Начать лечение гипогликемии тем же путем. Измерить уровень глюкозы через 15 - 20 минут после начала лечения, если результатов нет - повторить лечение. Для пациентов без сознания и/или с судорогами (если невозможен пероральный прием глюкозы) - внутривенное введение 10% декстрозы** в дозе 5 мл/кг. Если эпизоды гипогликемии повторяются - сообщите диетологу, возможно нужно изменить кетосоотношение.
III. Кетоз 
Провести исследование кислотно-основного состояния и газов крови. Если pH нормальный - продолжайте КД. Если pH  (некомпенсированный ацидоз
(некомпенсированный ацидоз  ) - снизить отношение при КД или выпить углеводный раствор (например, 20 - 50 мл фруктового сока).
) - снизить отношение при КД или выпить углеводный раствор (например, 20 - 50 мл фруктового сока).
Лечение гиперкетонемии. Включение в диету или внутривенное введение дополнительных углеводов:
- 20 - 50 мл фруктового сока
- 1 чайная ложка сахара, варенье, мед или сироп (можно с водой, чаем)
- Повторное определения кетонов в крови через 120 минут после приема дополнительных углеводов
- Если кетонемия >= 6 ммоль/л и/или определяются симптомы ацидоза - дайте ребенку еще 30 мл сока/чая с сахаром
- Если вторая порция углеводов не имеет эффекта, то необходимо внутривенное введение 5% раствора декстрозы**
- Контроль уровня кетонов каждые 6 часов
- Нет необходимости в контроле гликемии. Необходимые показатели более 2,6 ммоль/л.
ЗАКЛЮЧЕНИЕ:
- При высоком кетозе и хорошем самочувствии - наблюдение
- При высоком кетозе и общей слабости - дополнительное обследование и лечение сопутствующих заболеваний
- Высокий кетоз и ацидоз - внутривенное введение 5% раствора декстрозы** и 0,9% раствора натрия хлорида**
- Высокий кетоз и гипогликемия - стандартное лечение гипогликемии.
Приложение Г1 - ГN
ШКАЛЫ
ОЦЕНКИ, ВОПРОСНИКИ И ДРУГИЕ ОЦЕНОЧНЫЕ ИНСТРУМЕНТЫ СОСТОЯНИЯ
ПАЦИЕНТА, ПРИВЕДЕННЫЕ В КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЯХ
Приложение Г1
СИМПТОМЫ, НАБЛЮДАЕМЫЕ ПРИ СИНДРОМЕ ДЕФИЦИТА GLUT1
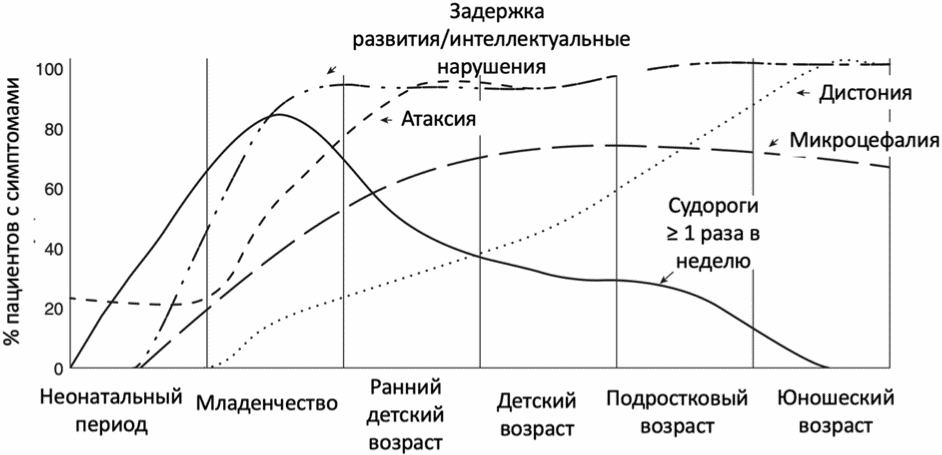
Рисунок 1 Симптомы, наблюдаемые при синдроме Glut1 в зависимости от возраста [39]
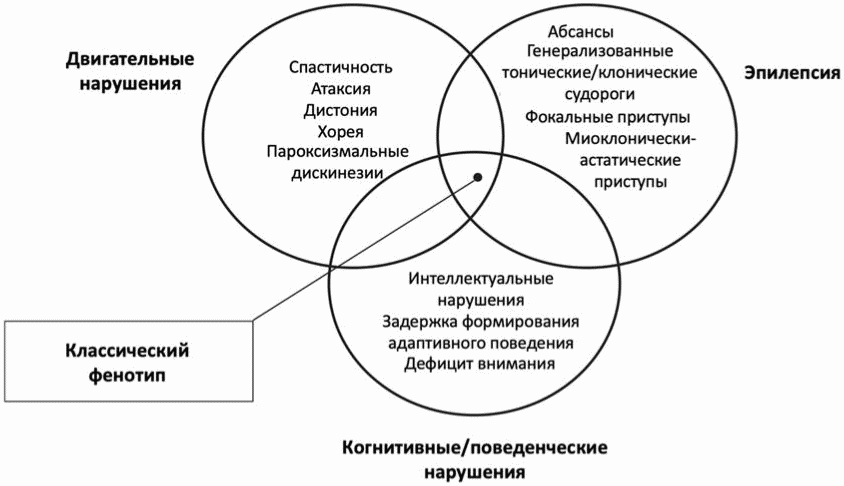
Рисунок 2 Неврологические проявления при синдроме дефицита Glut1 [45]
Приложение Г2
ГЕНОТИП-ФЕНОТИПИЧЕСКАЯ КОРРЕЛЯЦИЯ
ПРИ СИНДРОМЕ ДЕФИЦИТА GLUT1
Была отмечена корреляция между конкретным типом патогенного варианта гена SLC2A1 и клинической тяжестью [18].
Пятьдесят три пациента были разделены клинически в соответствии с неврологической шкалой Колумбийского университета (CNS - Columbia Neurological Score) на четыре группы [18]:
- Минимальная (70 - 76 баллов)
- Легкая (60 - 69 баллов)
- Умеренная (50 - 59 баллов)
- Тяжелая (40 - 49 баллов)
Сравнение типов гетерозиготных патогенных вариантов в гене SLC2A1 среди четырех групп выявило следующее:
- Миссенс-варианты встречались преимущественно при легкой и умеренной тяжести неврологических нарушений.
- Варианты сайтов сплайсинга, нонсенс-варианты, инсерции, делеции экзонов встречались практически всегда при средней и тяжелой степени неврологических нарушений.
- Полные делеции генов наблюдались при тяжелой степени.
Значимая обратная корреляция (R2 = 0,94) наблюдалась между средними значениями поглощения 3-O-метил-D-глюкозы эритроцитами и клинической тяжестью, определенной по неврологической шкале Колумбийского университета. Таким образом, поглощение глюкозы эритроцитами является показателем функционального эффекта патогенного варианта.
Было идентифицировано множество патогенных вариантов [18, 69]. Было обнаружено несколько горячих точек мутаций и участков генов:
- Три человека (каждый из разных семей) имеют мягкий клинический фенотип (определяемый по шкале CNS) и гетерозиготный p.Arg333Trp патогенный миссенс-вариант.
- Двенадцать человек имеют гетерозиготный патогенный миссенс-вариант по аминокислотному остатку аргинин 126.
- Пятеро имели мягкий фенотип: трое членов одной семьи с патогенным вариантом p.Arg126His и два члена одной семьи с патогенным вариантом p.Arg126Cys.
- Пять с классическим фенотипом или неэпилептическим фенотипом имели патогенный вариант p.Arg126Cys.
- Два монозиготных брата-близнеца с пароксизмальным хореоатетозом со спастичностью были гетерозиготными по патогенному варианту p.Arg126Cys.
- Значительная часть (5/21) патогенных вариантов расположена в уязвимой области белка Glut1, который включает четвертый трансмембранный домен, кодируемый экзоном 4, что указывает на критическое функциональное нарушение, связанное со структурными изменениями в этой области белка
- Три человека были гетерозиготами по патогенному варианту p.Thr295Met:
- Классический фенотип с ранним началом, включающий ежемесячные приступы, начинающиеся в младенчестве, задержку развития, атаксию, микроцефалию и языковой дефицит;
- Гипогликоррахия и низкие нормальные концентрации лактата в спинномозговой жидкости;
- Нормальный приток 3-OMG (3-O-methyl-d-glucose) в эритроциты.
Исследования мутагенеза показывают, что p.Thr295Met специфически влияет на поток глюкозы, нарушая отток, а не приток. Эти находки объясняют кажущиеся парадоксальными находки синдрома дефицита Glut1 с гипогликоррахией и "нормальным" поглощением глюкозы эритроцитами [68].
Приложение Г3
НЕОБХОДИМЫЕ ОБСЛЕДОВАНИЯ ПЕРЕД ВВЕДЕНИЕМ В КД
Осмотр пациента перед началом кетогенной диеты:
- Характеристика судорог (Тип, частота)
- Сопутствующие болезни
- Препараты, постоянно принимающиеся на момент осмотра
- Привычки в еде
- Непереносимость определенных продуктов
- Оценка соматического статуса
- Оценка неврологического статуса
- Антропометрические исследования (измерение массы тела, роста, окружности головы)
Необходимые обследования перед введением в КД
|
Исследование (базовое)
|
|
|
Обязательные
|
Рекомендуемые
|
|
Кровь:
- Общий (клинический) анализ крови развернутый
- Анализ крови биохимический общетерапевтический (исследование уровня общего белка, альбумина, мочевины, креатинина, триглицеридов, холестерина, общего билирубина, билирубина связанного (конъюгированного), билирубина свободного (неконъюгированного), глюкозы, щелочной фосфатазы, холестерина липопротеинов низкой плотности, холестерина липопротеинов высокой плотности, натрия, калия, общего магния, ионизированного магния, общего кальция, ионизированного кальция, 25-OH витамина Д, хлоридов, мочевой кислоты, креатинкиназы, активности аспартатаминотрансферазы, активности аланинаминотрансферазы, активности амилазы
- Исследование кислотно-основного состояния и газов крови
- Общий (клинический) анализ мочи
- Исследование уровня фосфора, уровня кальция, уровня мочевой кислоты, уровня креатинина, уровня калия, уровня натрия, кетоновых тел, уровня хлоридов в моче
Функциональная диагностика:
- Электроэнцефалограмма
- Ультразвуковое исследование органов брюшной полости (комплексное) и почек
|
Кровь:
- определение уровня витамина B12 (цианокобаламина), цинка, селена
- исследование уровня ферритина в крови
- исследование уровня свободного карнитина и ацилкарнитинов в крови
|
Приложение Г4
КЕТОГЕННАЯ ДИЕТА: ПОКАЗАНИЯ И ПРОТИВОПОКАЗАНИЯ
|
Показания
|
Противопоказания
|
|
Эпилепсия:
- рефрактерная эпилепсия после применения двух противоэпилептических препаратов
- синдром Веста
- синдром Отахара
- фокальные судороги в период ожидания хирургического лечения эпилепсии
|
Абсолютные:
- нарушение окисления жирных кислот (VLCAD, LCHAD, MCAD, OCTN2, CPT1, CPT2)
- дефицит пируваткарбоксилазы и другие дефекты гликонеогенеза (дефицит фруктозо-1,6-дифосфата)
- дефекты кетолиза
- дефекты кетогенеза
- порфирия
- синдром удлиненного интервала QT
|
|
Метаболические заболевания:
- синдром дефицита Glut1
- дефицит PDHC
- митохондриальные нарушения
|
Относительные:
- невозможность обеспечения полноценного питания
- очаг, требующий хирургического вмешательства, выявляемый при нейровизуализации и видео-ЭКГ-мониторинге
- несоблюдение назначений родителями или лицами, осуществляющими уход за ребенком
|
Приложение Г5
СООТНОШЕНИЕ
МАКРОНУТРИЕНОВ ПРИ РАЗЛИЧНЫХ ВАРИАНТАХ КД,
РАССЧИТАННЫХ НА РАЦИОН 1200 ККАЛ
|
Макронутриенты
|
4:1 КД
|
3:1 КД
|
2:1 КД
|
1:1 КД
|
MCT КД
|
МДА
|
|
Жиры (г)
|
120
|
116
|
109
|
97
|
67 - MCT
28 - LCT
|
87
|
|
Жиры (%)
|
90
|
87
|
82
|
70
|
50 - MCT
21 - LCT
|
65
|
|
Белки (г)
|
20.1
|
30
|
35
|
46
|
57
|
90
|
|
Белки (%)
|
6.7
|
10
|
12
|
15
|
19
|
29 - 32
|
|
Углеводы (г)
|
9.9
|
19
|
19
|
46
|
30
|
15
|
|
Углеводы (%)
|
3.3
|
6
|
6
|
15
|
10
|
3 - 6
|
Приложение Г6
СРАВНЕНИЕ
РАЗЛИЧНЫХ ВАРИАНТОВ КД, ПРИМЕНЯЕМЫХ ПРИ ДЕФИЦИТЕ GLUT1
|
Вопросы
|
КД и Модифицированная КД
|
MCT КД
|
МДА
|
|
Диета вводится под медицинским наблюдением?
|
да
|
да
|
да
|
|
Это высокожировая диета?
|
да
|
да
|
да
|
|
Это низкоуглеводная диета?
|
да
|
да
|
да
|
|
Какое соотношение макронатуриентов: жиры:белки + углеводы используется?
|
4:1, 3:1, 2:1, 1:1
|
Примерно 1:1
|
Примерно 2:1
|
|
Какое количество углеводов разрешается, если калорийность рациона 1000 ккал?
|
8 г - 4:1
16 г - 3:1
30 г - 2:1
40 - 60 г - 1:1
|
40 - 50 г
|
10 - 15 г - подростки
20 г - взрослые
|
|
Где происходит введение в диету
|
В больнице или дома под наблюдением врача
|
В больнице или дома под наблюдением врача
|
дома
|
|
Происходит расчет калорий?
|
да
|
да
|
При необходимости
|
|
Необходимо включать в терапию поливитамины с минеральными веществами?
|
да
|
да
|
да
|
|
Есть ли ограничения по жидкости?
|
нет
|
нет
|
нет
|
|
Проводится ли медицинское обследование перед началом диеты?
|
да
|
да
|
да
|
|
Могут ли быть побочные эффекты?
|
да
|
да
|
да
|
Приложение Г7
СРЕДНЕСУТОЧНЫЕ НОРМЫ
ФИЗИОЛОГИЧЕСКИХ ПОТРЕБНОСТЕЙ В ЭНЕРГИИ ДЛЯ ДЕТЕЙ
Среднесуточные нормы физиологических потребностей в энергии для детей согласно FAO/WHO/UNU 2001
|
Возраст (мес.)
|
Энергия (ккал/кг) муж
|
Энергия (ккал/кг) жен
|
|
0 - 1
|
113
|
107
|
|
1 - 2
|
104
|
101
|
|
2 - 3
|
95
|
94
|
|
3 - 4
|
82
|
84
|
|
4 - 5
|
81
|
83
|
|
5 - 6
|
81
|
82
|
|
6 - 7
|
79
|
78
|
|
7 - 8
|
79
|
78
|
|
8 - 9
|
79
|
78
|
|
9 - 10
|
80
|
79
|
|
10 - 11
|
80
|
79
|
|
11 - 12
|
81
|
79
|
|
Возраст (годы)
|
||
|
1 - 2
|
82,4
|
80,1
|
|
2 - 3
|
83,6
|
80,6
|
|
3 - 4
|
79,7
|
76,5
|
|
4 - 5
|
76,8
|
73,9
|
|
5 - 6
|
74,5
|
71,5
|
|
6 - 7
|
72,6
|
69,3
|
|
7 - 8
|
70,5
|
66,7
|
|
8 - 9
|
68,5
|
63,8
|
|
9 - 10
|
66,6
|
60,8
|
|
10 - 11
|
64,6
|
57,8
|
|
11 - 12
|
62,4
|
54,8
|
|
12 - 13
|
60,2
|
52,0
|
|
13 - 14
|
57,9
|
49,3
|
|
14 - 15
|
55,6
|
47,0
|
|
15 - 16
|
53,4
|
45,3
|
|
16 - 17
|
51,6
|
44,4
|
|
17 - 18
|
50,3
|
44,1
|
Энергетические потребности детей раннего возраста, получающих КД, на основе RDA <a>
|
Возраст (месяцы)
|
Вес (кг)
|
Ккал/кг/день
|
|
1 - 3
|
3,8 - 5,9
|
100 - 95
|
|
4 - 6
|
6,0 - 7,9
|
95 - 85
|
|
7 - 12
|
8,0 - 10,0
|
85 - 80
|
--------------------------------
Примечание:
<a> На основе среднего показателя для Австралии, Франции, Германии, Нидерландов, Великобритании, США.
Приложение Г8
РЕКОМЕНДУЕМАЯ ЕЖЕДНЕВНАЯ ПОТРЕБНОСТЬ
В БЕЛКАХ У ПАЦИЕНТОВ, ПОЛУЧАЮЩИХ КД
На основе значений RDA
|
Возраст
|
норма белка г/кг/день
|
|
0 - 12 мес.
|
1,5
|
|
13 - 35 мес.
|
1,1
|
|
3 года
|
1,1
|
|
4 - 13 лет
|
0,95
|
|
14 - 18 лет
|
0,85
|
|
18 лет
|
0,8
|
На основе значений RDA или WHO/FAO
|
Возраст (месяцы)
|
Вес, кг (с учетом идеального соотношения вес/возраст)
|
Белки г/кг/день в период КД <a>
|
белки/г/кг/день WHO/FAO
|
|
1 - 3
|
3,8 - 5,9
|
2,0 - 1,6
|
1,77 - 1,36 <b>
|
|
4 - 6
|
6,0 - 7,9
|
1,5 - 1,3
|
1,24 - 1,12 <b>
|
|
7 - 12
|
8,0 - 10,0
|
1,2 - 1,1
|
1,12 - 0,86 <c>
|
--------------------------------
Примечание:
<a> - На основе среднего показателя для Австралии, Франции, Германии, Нидерландов, Великобритании, США;
<b> - WHO 2007: безопасный уровень для роста +1.96 SD;
<c> - FAO 2013.
Приложение Г9
РЕКОМЕНДУЕМОЕ ЕЖЕДНЕВНОЕ ПОТРЕБЛЕНИЕ ЖИДКОСТИ
|
Вес (кг)
|
Потребность в жидкости
|
|
1 - 10
|
100 мл/кг
|
|
11 - 20
|
1000 мл + (50 мл/кг на каждый кг > 10 кг)
|
|
> 20
|
1500 мл (20 мл/кг на каждый кг > 20 кг)
|
Рекомендуемое ежедневное количество жидкости на основе RDA <a>
|
Возраст (месяцы)
|
Вес (кг)
|
мл/кг/день
|
|
1 - 3
|
3,8 - 5,9
|
150 - 140
|
|
4 - 6
|
6,0 - 7,9
|
120 - 110
|
|
7 - 12
|
8,0 - 10,0
|
100 - 90
|
--------------------------------
Примечание:
<a> - На основе среднего показателя для Австралии, Франции, Германии, Нидерландов, Великобритании, США.
Приложение Г10
БЕЗОПАСНЫЕ УРОВНИ
ПОТРЕБЛЕНИЯ БЕЛКА И ЭНЕРГЕТИЧЕСКИЕ ПОТРЕБНОСТИ
У ДЕТЕЙ И ВЗРОСЛЫХ, А ТАКЖЕ В ПЕРИОД БЕРЕМЕННОСТИ
И ЛАКТАЦИИ (ВЫБОРОЧНО ИЗ РЕКОМЕНДАЦИЙ FAO/WHO/UNU
ДЛЯ ЗДОРОВОЙ ПОПУЛЯЦИИ)
|
Потребление белка
|
|||||||
|
Возраст
|
Потребление
|
Возраст
|
Женщины
|
Мужчины
|
женщины
|
мужчины
|
|
|
Месяцы
|
г/кг веса тела в день
|
годы
|
кДж/кг веса тела в день
|
ккал/кг веса тела в день
|
|||
|
1
|
1,77
|
0,5
|
340
|
35
|
81,3
|
80,0
|
|
|
2
|
1,50
|
2,5
|
334
|
48
|
79,8
|
83,2
|
|
|
3
|
1,36
|
5,0
|
305
|
15
|
72,9
|
75,3
|
|
|
6
|
1,31
|
10
|
248
|
75
|
59,3
|
65,7
|
|
|
12
|
1,14
|
15
|
193
|
30
|
46,1
|
55,0
|
|
|
1,5
|
1,03
|
||||||
|
2
|
0,97
|
||||||
|
3
|
0,90
|
18 - 29
|
159
|
83
|
38,0
|
43,7
|
|
|
4 - 6
|
0,87
|
30 - 59
|
148
|
75
|
35,4
|
41,8
|
|
|
7 - 10
|
0,92
|
||||||
|
женщины
|
мужчины
|
||||||
|
Годы
|
|||||||
|
11
|
0,90
|
0,91
|
18 - 29
|
180
|
12
|
43,0
|
50,7
|
|
12
|
0,89
|
0,90
|
30 - 59
|
183
|
12
|
43,7
|
|
|
13
|
0,88
|
0,90
|
|||||
|
14
|
0,87
|
0,89
|
|||||
|
15
|
0,85
|
0,88
|
Триместр
|
кДж/день
|
ккал/день
|
||
|
16
|
0,84
|
0,87
|
1
|
375
|
90
|
||
|
17
|
0,83
|
0,86
|
2
|
1200
|
287
|
||
|
18
|
0,82
|
0,85
|
3
|
1950
|
466
|
||
|
> 18
|
0,83
|
0,83
|
|||||
|
Беременность
|
Месяцы
|
кДж/день
|
ккал/день
|
||||
|
Общее дополнительное потребление белка
|
1 - 6
|
2800
|
669
|
||||
|
> 6
|
1925
|
460
|
|||||
|
Триместр
|
г/день
|
||||||
|
1
|
1
|
||||||
|
2
|
10
|
||||||
|
3
|
31
|
||||||
|
Лактация
|
|||||||
|
Общее дополнительное потребление белка
|
|||||||
|
месяцы
|
г/день
|
||||||
|
0 - 6
|
19
|
||||||
|
> 6 - 12
|
13
|
||||||
Приложение Г11
ПОДГОТОВИТЕЛЬНЫЙ, НАЧАЛЬНЫЙ И ОСНОВНОЙ ЭТАПЫ
ЛЕЧЕНИЯ НА ОСНОВЕ КЕТОГЕННОЙ ДИЕТЫ У ДЕТЕЙ РАННЕГО ВОЗРАСТА
|
Подготовка
|
Начало
|
Терапевтическая фаза
|
|
Исключить противопоказания
(Прил. Г4)
|
Начальное соотношение - 1:1
(возможно сразу 3:1),
целевое - 3:1
(1:1 - > 2:1 - > 3:1)
|
Эффективность КД: дневник приступов (при необходимости ЭЭГ)
|
|
Подготовка к лечению
(Прил. Г3)
|
Переносимость КД: ЖКТ, сон, поведение, аппетит
|
|
|
Учитывать потребности питания
- Калории (Прил. Г7)
- Белки (Прил. Г8)
- Жидкости (Прил. Г9)
|
||
|
Клинический мониторинг: (Прил. Г12)
|
||
|
УЗИ почек: через 6 - 12 месяцев
|
||
|
Обследование до начала терапии (Прил. 3)
|
||
|
Корректировка
Исследование уровня глюкозы в крови: дважды в день
Обнаружение кетоновых тел в моче: дважды в день
|
||
|
Клиническая и параклиническая оценка на начальном и основном этапах диетотерапии
|
||
|
Гипогликемия
|
Кетоз
|
|
|
Тревожность плохой тонус, сонливость, бледность, низкая температура, холодное и влажное тело, цианоз
|
Учащенное дыхание, учащенное сердцебиение, покраснение лица, раздражительность, рвота, сонливость, проблемы с кормлением
|
|
Приложение Г12
РЕКОМЕНДУЕМЫЙ КОНТРОЛЬНЫЙ КЛИНИЧЕСКИЙ МОНИТОРИНГ
ПРИ СОБЛЮДЕНИИ КЕТОГЕННОЙ ДИЕТЫ
|
Обследования
|
Частота мониторинга
|
|
Оценка эффективности КД путем изучения дневника приступов, частоты применения лекарственных препаратов, либо анализа других показателей возможных улучшений
|
При каждом контрольном визите
|
|
Оценка переносимости КД и побочных эффектов, включая вопросы о функции кишечника, сне, поведении и аппетите
|
При каждом контрольном визите
|
|
Вес, рост, окружность головы
|
При каждом контрольном визите
|
|
Общий (клинический) анализ крови развернутый
Общий (клинический) анализ мочи
|
Как указано в Прил. Г13. Частоту анализов повышают при выявлении отклонений или наличии других нежелательных явлений
|
|
Ультразвуковое исследование органов брюшной полости (комплексное)
|
Через 6 - 12 месяцев на КД. Дополнительное обследование при клинических показаниях
|
|
Ультразвуковое исследование почек
|
Через 6 - 12 месяцев на КД. Дополнительное обследование при клинических показаниях в виде гематурии в 3 последовательных тестах или необъяснимой раздражительности у ребенка (на начальном и основном этапах КД)
|
|
Электроэнцефалограмма
|
По клиническим показаниям
|
|
Электрокардиограмма
|
До начала лечения и по клиническим показаниям
|
Приложение Г13
РЕКОМЕНДУЕМЫЙ ЛАБОРАТОРНЫЙ МОНИТОРИНГ В ПЕРИОД ДИЕТОТЕРАПИИ
|
Исследования
|
Частота мониторинга
|
|
Обязательные:
|
|
|
Кровь:
|
|
|
Общий анализ крови
|
через 3 месяца, 6 месяцев, затем каждые 6 месяцев
|
|
Почечный профиль (натрий, калий, мочевина, бикарбонат, альбумин)
|
|
|
Печеночный профиль
|
|
|
Кальций, фосфат, магний
|
|
|
Глюкоза
|
|
|
Витамин D
|
каждые 6 месяцев
|
|
Исследование уровня холестерина, уровня холестерина липопротеинов низкой плотности в крови, уровня липопротеинов в крови
|
через 3 месяца, 6 месяцев, затем каждые 6 месяцев
|
|
Моча:
|
|
|
Соотношение кальций:креатинин, гематурия
|
через 3 месяца, 6 месяцев, затем каждые 6 месяцев
|
|
Рекомендованные (по возможности):
|
|
|
Кровь:
|
|
|
Определение уровня витамина B12 (цианокобаламин) в крови
Исследование уровня фолиевой кислоты в сыворотке крови
|
по показаниям
|
|
Исследование уровня цинка в крови
Исследование уровня селена в крови
|
|
|
Исследование уровня ферритина в крови
|
|
|
Свободный карнитин и профиль ацилкарнитинов
|
Приложение Г14
ПОБОЧНЫЕ ЭФФЕКТЫ КД, ИХ ПРОФИЛАКТИКА И КОРРЕКЦИЯ
|
Осложнения
|
Профилактика
|
Методы коррекции
|
|
Гипогликемия
|
Прогредиентное введение диеты, мониторинг глюкозы в крови
|
Прием 5 г углеводов (раствора декстрозы**, фруктового сока - 50 мл, молока - 100 мл)
|
|
Гиперкетонемия
|
Мониторинг уровня кетоновых тел, КОС, своевременная коррекция рациона питания
|
Прием 5 г углеводов (раствора декстрозы**, фруктового сока - 50 мл, молока - 100 мл)
|
|
Гастроинтестинальные нарушения: рвота, диарея
|
Прием курсами препаратов ферментов поджелудочной железы, желчегонных средств
|
Уменьшение количества жиров в рационе (на 30 - 50%) в зависимости от тяжести симптоматики:
- Стимуляторы моторики желудочно-кишечного тракта
- Препараты, способствующие пищеварению, включая ферментные препараты
- Препараты для лечения заболеваний желчевыводящих путей
- регидратация
- симптоматическая терапия
|
|
Со стороны мочевыделительной системы: гиперуратурия, гиперкальциурия
|
Соблюдение питьевого режима, контроль клинических и биохимических параметров мочи
|
Пациентам старше 12 лет #Калия гидрокарбонат + Лимонная кислота + Натрия цитрат; лимонного сока (до 85 г в сутки); терапия с учетом результатов исследования мочи
|
|
Изменения со стороны сердечно-сосудистой системы (удлинение интервала QT, дилатационная кардиомиопатия)
|
Дополнительное назначение препаратов селена. Мониторирование электрокардиографических данных
|
Препараты калия, #убидекаренон
|
Приложение Г15
ФОРМУЛЫ
ШОФИЛДА ДЛЯ РАСЧЕТА ВЕЛИЧИНЫ ОСНОВНОГО ОБМЕНА
ПО МАССЕ ТЕЛА У ДЕТЕЙ
|
Возраст и пол
|
Формула
|
Стандартная ошибка вычисления
|
|
До 3 лет
|
||
|
Мальчики
|
ВОО <*> (МДж/сут) = 0,249 x МТ <**> - 0,127
|
0,293
|
|
Девочки
|
ВОО (МДж/сут) = 0,244 x МТ - 0,130
|
0,246
|
|
Мальчики
|
ВОО (ккал/сут) = 59,5 x МТ - 30,4
|
70
|
|
Девочки
|
ВОО (ккал/сут) = 58,3 x МТ - 31,1
|
59
|
|
3 - 10 лет
|
||
|
Мальчики
|
ВОО (МДж/сут) = 0,095 x МТ + 2,110
|
0,280
|
|
Девочки
|
ВОО (МДж/сут) = 0,085 x МТ + 2,033
|
0,292
|
|
Мальчики
|
ВОО (ккал/сут) = 22,7 x МТ + 504,3
|
67
|
|
Девочки
|
ВОО (ккал/сут) = 20,3 x МТ + 485,9
|
70
|
|
10 - 17 лет
|
||
|
Мальчики
|
ВОО (МДж/сут) = 0,074 x МТ + 2,754
|
0,440
|
|
Девочки
|
ВОО (МДж/сут) = 0,056 x МТ + 2,898
|
0,466
|
|
Мальчики
|
ВОО (ккал/сут) = 17,7 x МТ + 658,2
|
105
|
|
Девочки
|
ВОО (ккал/сут) = 13,4 x МТ + 692,6
|
111
|
--------------------------------
Примечание:
<*> ВОО - величина основного обмена,
<**> МТ - масса тела (кг).
Приложение Г16
ПРОДУКТЫ И МАСЛА, ИСПОЛЬЗУЕМЫЕ ПРИ КД
|
Название
|
Описание
|
Состав
|
|
КЕТОКАЛ 3:1
(Nutricia)
|
Специализированный продукт детского диетического лечебного питания при лекарственно-резистентной эпилепсии и других состояниях, при которых показана кетогенная диета, сухая смесь с нейтральным вкусом
|
Смесь растительных масел, молочные белки (казеин и сывороточные белки), а также аминокислоты, углеводы, витамины, минералы и микроэлементы
|
|
КЕТОКАЛ 4:1
(Nutricia)
|
Специализированный продукт детского диетического лечебного питания при лекарственно-резистентной эпилепсии и других состояниях, при которых показана кетогенная диета, сухая смесь с нейтральным вкусом
|
Смесь растительных масел, молочные белки (казеин и сывороточные белки), а также аминокислоты, углеводы, витамины, минералы, микроэлементы и пищевые волокна
|
|
Ликвиджен****
(Nutricia)
|
Энергетический модуль, высококалорийная эмульсия среднецепочечных триглицеридов
|
MCT (Кокосовое масло фракционированное, пальмовое масло фракционированное), Вода деминерализованная, Эмульгатор (E472c)
|